KEYWORDS
Arthralgia, haemochromatosis, iron, transferrin receptor 2
INTRODUCTION
Hereditary haemochromatosis (HH) is a genetic disease characterised by body iron loading. If not recognised and treated early, the excess iron may lead to complications such as arthropathy, liver disease, diabetes and cardiomyopathy.1 The most common form, type 1 HH, is caused by mutations in the HFE gene.2 However, mutations in HJV, HAMP, TFR2 and SLC40A1 can also cause HH: types 2A, 2B, 3 and 4(A and B), respectively. All these types have an autosomal recessive inheritance, except for type 4A and B, which are autosomal dominant. The HH genes are related to regulation of circulating and tissue iron levels by the hepcidin-ferroportin axis. The hormone hepcidin normally inhibits iron uptake from the gut and iron release from the reticuloendothelial system by promoting degradation of the cellular iron exporter ferroportin.3
Five different HH disorders may lead to a classical HH phenotype, i.e. normal haemoglobin (Hb), elevated ferritin and transferrin saturation (TSAT) and iron overload in the parenchymal tissues, such as the liver and pancreas. The pathophysiology of these five conditions is similar: inadequate or ineffective hepcidin-mediated downregulation of ferroportin and subsequent increased iron absorption from the diet, relatively low iron content in the reticuloendothelial system and high parenchymal and circulating iron levels.
HFE-related HH is by far the most common form of HH, and its prevalence is mainly restricted to patients from northern European descent.4 Most patients with HFE-HH do not present until middle age (and women not until after menopause). The other HH subtypes are more rare and less restricted to certain populations.5,6 Juvenile forms of HH are due to mutations in the genes encoding for haemojuvelin (HJV, HH type 2A) and hepcidin (HAMP, HH type 2B). These forms of HH are generally characterised by their early onset and a particularly severe phenotype, with patients typically presenting before the age of 30 years with severe systemic iron overload, heart failure and endocrine disorders.7
Another form of classical HH is caused by mutations in SLC40A1 (ferroportin) that interfere with the regulation by hepcidin.8,9 Since this causes excessive ferroportinmediated intestinal iron uptake, these mutations are described as gain-of-function mutations. The phenotype of this so-called HH type 4B in affected patients is similar to that in patients with HFE-HH.10,11
A more atypical form of HH is caused by loss-offunction ferroportin mutations, a condition referred to as ‘ferroportin disease’,8 or type 4A HH. Iron overload in this disease is restricted to the reticuloendothelial system, which leads to the combination of high ferritin concentrations, normal TSAT, and in rare cases iron-restricted erythropoiesis with mild anaemia or impaired tolerance of phlebotomy.12 Patients with type 4A rarely develop iron-related disease symptoms.
Type 3 HH (TFR2-related) is rare and more severe than type 1 HH and presents in young adults.1,2 Patients typically present in young adulthood, although some children with type 3 HH have been reported as well. Clinical symptoms are similar to those in type 1 and are also treated by phlebotomy. TFR2 variants have been reported in various populations, most prominently in Italian and Asian families.13
Around 50 patients with type 3 HH have been described,14 comprising around 50 (possibly) pathogenic mutations identified so far.6,15 The first two type 3 HH families were identified in 2000, when the function of TFR2 was still unknown.16 A few years later, it was discovered that mutants of this protein cause down-regulation of hepcidin.17 Current evidence supports a model in which TFR2 senses high levels of circulating iron by its binding to diferric transferrin18 and subsequently interacts with HFE on the membrane of the hepatocyte, which leads to a SMAD-mediated intracellular signalling resulting in elevated hepcidin expression.19 Since this upregulation is lost in type 3 HH, hepcidin levels are inappropriately low for circulating iron levels and tissue iron stores, resulting in excessive dietary iron absorption via ferroportin.
We describe three women and one man from three families diagnosed with HH type 3 in our haemochromatosis referral centre in the Netherlands who presented with iron loading and arthralgia in young adulthood. During their diagnostic process we identified in total four different pathogenic TFR2 variants, of which three are novel. With this report, we aim to facilitate early diagnosis of type 3 HH and related complications by increasing the knowledge and awareness of the disease among physicians.
MATERIALS AND METHODS
Patients
We retrospectively collected and reviewed data on clinical presentation, biochemical tests and DNA analysis of four patients who were diagnosed with type 3 HH at the Radboudumc between 2009 and 2016.
Laboratory methods
Genotyping was performed by DNA sequence analysis of the full coding part of the genes by Sanger sequencing, or whole-exome sequencing (WES), depending on the patient. In case of WES, we used our diagnostic gene panel ‘Iron disorders’ – consisting of 44 genes with reported roles in iron metabolism disorders – as a filter to analyse the exons of iron-related genes only.20 The pathogenicity of the variants found was assessed by association of the variant with the phenotype within a family, in silico tools and review of the literature and variant databases (ExAC Browser and dbSNP).21,22 The in silico analysis was performed with Alamut Visual (Interactive Biosoftware), which comprises several predictive programmes to assess the consequence of missense and splice site mutations.23 In case of missense mutations, we also used HOPE (Have (y) Our Protein Explained) to get an in silico prediction of the functional consequences of the mutation.24
Serum hepcidin-25 measurements were performed with weak cation exchange time of flight mass spectrometry (WCX-TOF MS), according to the method of Kroot et al.25 The serum hepcidin-25 (hepcidin) and hepcidin/ferritin ratios were interpreted in the context of the reference ranges in the general population.26
CASE SUMMARIES
Patient 1
Patient 1 is a woman of British origin, who presented with fatigue at the age of 25. The patient’s blood counts (leukocytes and platelets) and serum chemistry profile (glucose, liver enzymes, albumin) were within the reference range. The iron laboratory parameters were as follows: Hb 8.4 mmol/l (= 13.5 g/dl, conversion factor 1.61), iron 56 μmol/l, TSAT 94% and serum ferritin 831 μg/l. Hepatitis, haematological and inflammatory diseases were excluded. There were no signs of metabolic syndrome. Liver biopsy showed mild architectural disturbance with bridging fibrosis between portal tracts, but no evidence of hepatitis or liver cirrhosis with an amount of 362.7 μmol iron per gram dry weight (= 20.25 mg/g; upper limit of reference in women < 36 μmol/g), which was consistent with haemochromatosis.27 The patient underwent 16 phlebotomies before she became pregnant at age 26. At 6 months of pregnancy, her ferritin was 440 μg/l and her TSAT was 100%. After her first pregnancy, at age 27, she presented with arthralgia of the pollex of her left hand and back pain. Phlebotomies were restarted after her second pregnancy at age 29, but due to lack of compliance, the patient’s serum ferritin and TSAT varied between 50-926 μg/l and 89-100%, respectively, between the ages of 27 and 40 years. At age 37, she also developed arthralgia of the metacarpophalangeal joints of digitus two, three and four of her right hand. X-ray of her hands demonstrated only mild degenerative changes of the carpal joints. Further investigation of the iron overload at age 40 showed a hepcidin/ferritin ratio of 1.6 pmol/μg (reference range premenopausal women 3.0-167.3 pmol/μg) (table 1).
When the patient was 29 years old, ferrokinetic studies with radioactive labelled iron were regularly used to obtain better insights into the defect of iron metabolism28-30 and were therefore also performed for our patient. After administration of Fe-59 bound to transferrin, plasma iron clearance, red cell iron incorporation and tissue iron uptake in time were assessed. Plasma T1/2 was prolonged to 186 min (normal 60-120 min).28 Iron incorporation after 14 days was low: 62.2% (reference 75-85%).28 Measurement of Fe-59 by a scintillation detector above the sacrum, heart, liver and spleen, 3 times a week for 2 weeks, showed a relatively high liver iron uptake. Finally, whole body counter measurements every 2 to 3 days for 2 weeks showed a body iron retention varying between 95.0% at day 2 to 96.5% at day 14, indicating hardly any body iron losses during this period of time. Taken together, in this patient, we observed a retarded plasma iron disappearance and incomplete iron incorporation in red blood cells, but increased iron uptake by the liver. This is in agreement with dilution of intravenously administrated iron by increased circulating iron levels and uptake of excess circulating iron by the hepatocytes.31 These observations corroborate the pathophysiology of HH, but nowadays do not have added diagnostic value.
DNA analysis revealed no mutations in the HFE gene. HAMP, HFE2 and SLC40A1 also revealed no pathogenic ions. Analysis of TFR2 by Sanger sequencing showed heterozygosity for the variants c.1606-2A>G in intron 13 and c.1870C>T (p.Gln624X) in exon 16. The c.1606-2A>G variant is present in the ExAC Browser at a frequency of < 1% (table 2) and was previously reported as ‘likely pathogenic but not previously reported among patients with HH’.6 Alamut Visual predicted the loss of a splice site at exon 14, which supports its suspected pathogenicity (table 2). Further investigation of the DNA of the patient’s mother revealed only the c.1606-2A>G variant on a single allele, confirming that both variants occur on two different alleles and thus that the patient is compound heterozygous for both TFR2 mutations. This confirmed the diagnosis for HH type 3. As the patient moved back to Britain, she was lost to follow-up.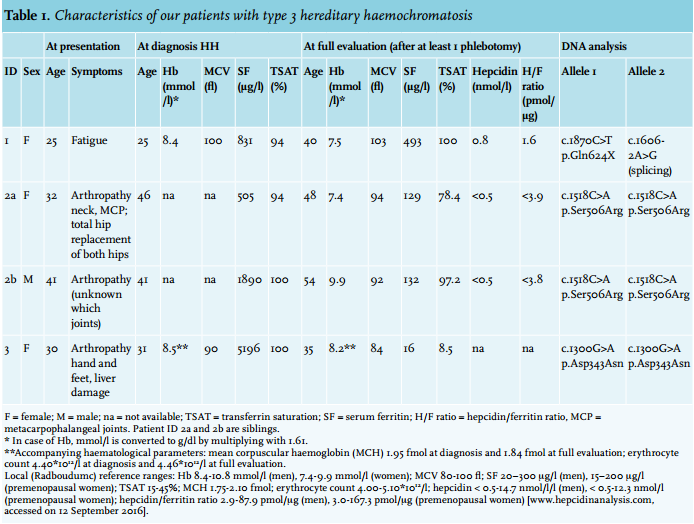
Patient 2a
Patient 2a is a 56-year-old woman of Dutch descent, who first presented elsewhere at age 32 with arthralgia in her neck and the metacarpophalangeal joints of digitus two and three of both hands. X-radiation of her hands, feet, ankles and knees revealed abnormalities consistent with haemochromatosis arthropathy. At the age of 45 years, she received a total hip replacement because of severe arthrosis, interpreted as a complication of familial hip dysplasia. The other hip was replaced two years later.
At the age of 46, she had a serum ferritin of 505 μg/l and a TSAT of 94%. The blood counts and her serum chemistry profile (glucose, liver enzymes, albumin) were within the reference range. Hepatitis, haematological and inflammatory diseases were excluded and there were no signs of metabolic syndrome. She was diagnosed with HH, yet screening of the HFE gene for the common p.Cys282Tyr and p.His63Asp variants was negative. She did not start phlebotomy treatment, as the iron loading was not considered to be severe: the arthrosis was thought to be independent of haemochromatosis. Two years after diagnosis, however, MRI of the liver was performed to further objectify iron overload and revealed an iron content of 250 μmol per gram dry weight (reference range 10-35 μmol/g).27
Subsequently, the patient started phlebotomy on an irregular basis and was also referred to our haemochromatosis referral centre for further analysis. A full evaluation revealed a serum hepcidin of < 0.5 nmol/l (reference range for premenopausal women < 0.5-12.3 nmol/l), which means it is below the detection limit for WCX-TOF MS, ferritin of 129 μg/l and a hepcidin/ferritin ratio of < 3.9 pmol/μg (table 1). Liver enzymes, α-fetoprotein, thyroid-stimulating hormone and glucose levels were all within the reference range. The rheumatologist diagnosed a nodular inflammatory polyarthrosis. Phlebotomy treatment was started to normalise ferritin levels to < 50 μg/l; unfortunately, it did not improve her arthralgia.
We found no pathogenic mutations in the HFE, HAMP, HFE2 and SLC40A1 genes. DNA sequencing analysis of TFR2, however, revealed a homozygous c.1518C>A transition changing a serine to an arginine at position 506 of the TFR2 protein (p.Ser506Arg). We found no notion of the pathogenicity of this mutation in the literature or variant databases (ExAC Browser, dbSNP).
In silico analysis predicted the variant to be pathogenic (table 2). Three-dimensional structure prediction analysis by HOPE showed that the mutation is located in the core of the protein and is not directly involved in ligand interaction or of importance for the dimerisation surface. However, because the arginine side chain is bigger than that of serine, the mutation likely leads to structural changes that prevent proper folding and thus function (figure 1).
Lastly, we performed restriction analysis for this newly discovered mutation in 100 randomly chosen controls. This revealed no detection of the p.Ser506Arg. In conclusion, the homozygous p.Ser506Arg mutation confirmed the diagnosis of type 3 HH.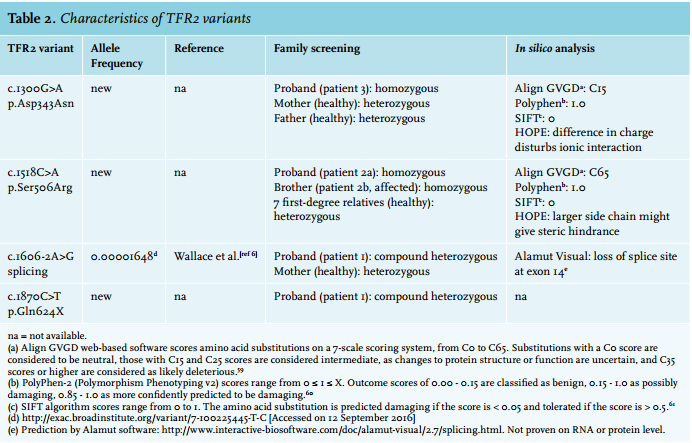

Patient 2b
Patient 2b is the brother of patient 2a and presented with severe arthrosis, an elevated serum ferritin level of 1890 μg/l and elevated TSAT at the age of 41 years. He was diagnosed with HH and treated with phlebotomies elsewhere, even though screening for HFE showed no mutations. A total of 50 phlebotomies were required to deplete his body iron stores to ferritin levels < 100 μg/l. This did not result in amelioration of his arthralgia. During regular, once every 8 weeks, maintenance phlebotomy, at 54 years of age and BMI 31.8 kg/m2 , his serum ferritin was 132 μg/l and TSAT was 97.2%. Further investigation showed that the serum hepcidin was < 0.5 nmol/l (reference range for men < 0.5-14.7 nmol/l) and the hepcidin/ferritin ratio was < 3.8 pmol/μg (reference range for men 2.9-87.9). The ALAT level was within the reference range.
After diagnosis of his sister, TFR2 DNA sequencing analysis was performed and he turned out to be homozygous for the novel p.Ser506Arg mutation as well. Subsequent family screening revealed heterozygosity for the mutation in all other tested family members. None of them showed a HH phenotype, which corroborates the supposed autosomal recessive inheritance.
Patient 3
Patient 3 is a woman who presented at age 30 with pre-eclampsia with hepatic dysfunction. After the delivery of a healthy son, one year later, she developed arthralgia in her hands and feet. Persistently mildly elevated liver enzymes (ASAT 51 U/l, reference 0-35 U/l; ALAT 83 U/l, reference 0-40 U/l) at this time warranted further examination, in which a ferritin level of 5196 μg/l and a TSAT of 100% were found, with Hb and other haematological parameters within reference ranges (table 1). Based on these values, she was diagnosed with HH. Extensive phlebotomy with a depletion phase of almost two years using a Port-A-Cath was initialised to decrease the ferritin levels to < 50 μg/l. Further analysis at our centre showed that this resulted in an apparent iron deficiency (ferritin 16 μg/l, TSAT 8.5%) without anaemia (Hb 8.2 mmol/l) (table 1). Furthermore, blood counts and glucose, but also liver enzymes were all within the reference range (ASAT 26 U/l; ALAT 21 U/l). Other problems in this patient known to be associated with iron overload included impaired fertility and hypothyroidism with subsequent myxoedema, attributed to Hashimoto’s disease.
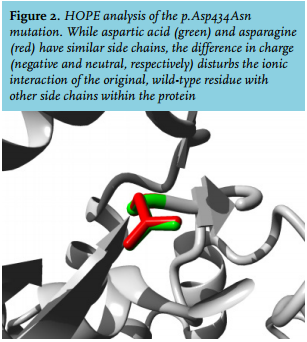
Screening of the HFE gene only showed the common heterozygous H63D mutation, which could not clarify the patient’s clinical phenotype. Subsequently, WES showed a homozygous c.1300G>A TFR2 variant, protein coding effect p.Asp434Asn. In silico analysis predicted this variant as likely pathogenic (table 2). Furthermore, HOPE analysis showed that the wild-type, aspartic acid residue normally forms a hydrogen bond with glycine at position 479 and aspartic acid at position 480 in the protein. It also forms salt bridges with arginine at position 165 and arginine at position 433. While the side chains of the wild-type (aspartic acid) and the mutant (asparagine) form have a similar structure (figure 2), the difference in charge (negative and neutral, respectively) disturbs the ionic interaction of the original, wild-type residue. Additionally, the substituted amino acid was highly conserved and part of the M28 peptidase domain, where other pathogenic mutations have also been identified.32
This variant has not been reported before in the literature or variant databases (ExAC database, dbSNP). Family screening showed that both parents were heterozygous carriers of the p.Asp434Asn mutation. This verified that this mutation was indeed present on two different alleles in the patient. Based on in silico analysis, frequency and family screening, we concluded that the p.Asp343Asn mutant is responsible for the patient’s clinical phenotype. Therefore, the diagnosis of HH type 3 was confirmed.
DISCUSSION
We describe four patients who presented with arthralgia at a relatively young age (27-41 years). Laboratory analysis showed elevated ferritin and TSAT levels. When measured, serum hepcidin turned out to be low relative to the ferritin concentration. Genetic testing eventually resulted in the detection of four different variants that are for the first time associated with the TFR2 defective phenotype. Furthermore, while early arthralgia is commonly found in both type 1 and type 3 HH,2,33 it is especially pronounced in our case series.
Although our sample number is small, it appears that while function is affected (arthropathy), survival is not. This is an important issue, because iron-related arthropathy can develop even when ferritin concentrations are moderately increased, as in patient 2a. This is in contrast to other symptoms, which are more likely to occur in individuals with serum concentrations of more than 1000 μg/l.34,35
A decreased hepcidin/ferritin ratio corroborates the pathophysiology of hereditary haemochromatosis.36-38 Indeed, in patient 1, the ratio was below the lower limit of the reference range. In patients 2a and 2b, it was < 3.9 pmol/μg and < 3.8 pmol/μg, respectively. The true value may be below the lower limit of the reference range, but this is unsure. In both patients, serum hepcidin was measured after phlebotomy treatment. As this removes iron from the body, it will further decrease the already low hepcidin levels. Moreover, increased erythropoiesis resulting from loss of erythrocytes also inhibits hepcidin production.39 These processes thus increase the chance that hepcidin levels drop below the detection limit. Therefore, to be able to draw any conclusions, our advice is to measure serum hepcidin levels before phlebotomy, when the hepcidin/ferritin ratio is more likely to be informative.
It is important to realise that while all type 3 HH patients have a low hepcidin/ferritin ratio, making it a sensitive marker, it is by no means a specific marker. For example, low ratios are also observed in some secondary forms of iron overload, such as aceruloplasminaemia and iron-loading anaemias,40-44 and the metabolic syndrome (Schaap CCM, Janssen MCH, Swinkels DW, unpublished findings). Iron loading anaemias include thalassaemia intermedia and sideroblastic anaemias, which lead to signs of iron overload even without transfusions. However, in contrast to HH patients, this overload is always accompanied by anaemia. A low hepcidin/ferritin ratio combined with a high TSAT and lack of anaemia therefore indicates HH type 1-3 as the most likely diagnosis.
In one of our patients, MRI was performed as recommended in the Dutch guidelines for patients with elevated ferritin and TSAT in the absence of the common HFE mutations.45 The liver iron content in this patient was 250 μmol/g dry weight, more than seven times the upper limit of the reference value (35 μmol/g dry weight).27 Several other diseases presenting with hyperferritinaemia, including alcoholic liver disease, hepatitis C infection, non-alcoholic fatty liver disease and liver cirrhosis, are also associated with increased hepatic iron deposition. However, in these diseases the liver iron content is typically below 100 μmol/g.46-48 Therefore, the high liver iron content quantified by MRI in our patient contributed to the diagnosis of HH. Importantly, the iron accumulation in HH is mainly parenchymal and therefore toxic, as indicated by high ferritin combined with high TSAT. For a certain degree of iron accumulation, parenchymal iron will not increase ferritin levels as much as iron accumulation in the reticuloendothelial system (which is for example observed in conditions with low-grade inflammation, such as metabolic syndrome or anaemia of chronic disease), as also demonstrated in transfusionindependent thalassaemia patients.49,50 Therefore, it should be kept in mind that in HH patients, liver iron content could reach toxic levels despite a relatively low ferritin.
When HH is suspected, it is useful to first test for the common p.Cys282Tyr and p.His63Asp variants in HFE. This is especially the case in patients of Northern European descent, since in these populations the prevalence of the p.Cys282Tyr variant is the highest, whereas in non-Caucasians the common HFE mutations are rare. In the absence of homozygosity for the p.Cys282Tyr variant and p.Cys282Tyr/p.His63Asp compound heterozygosity, several causative genes remain. Testing these genes (i.e. HJV, HAMP, TFR2 and SLC40A1) consecutively can be a time-consuming and costly process. With the recent developments in genetics, it may be advantageous to use techniques that screen for multiple genes at once. One suitable technique for this is WES, where the exons of all genes are sequenced. Using a diagnostic filter, genes involved in hereditary haemochromatosis and related diseases can easily be screened for mutations. While more expensive than testing for one gene, WES becomes cost-effective when testing three or more genes, and its costs are rapidly decreasing. We therefore recommend to screen for the common variants in HFE first. If the results do not explain the observed phenotype, WES should be considered. Of note, if causative variants are present in non-coding regions of the genome, they will not be detected by WES.
Once type 3 HH is diagnosed, the treatment of choice is phlebotomy, similar to that of type I HH.51 In the absence of randomised controlled trials, recommendations are based upon the clinical evidence that iron removal before onset of cirrhosis and diabetes in type I HH is associated with reduced morbidity and mortality.52,53
However, since solid evidence on the optimal endpoint of initial venesection and optimal maintenance therapy is lacking, worldwide guidelines are not identical at this point. According to the European guidelines, the goal is to reduce serum ferritin levels below 50 μg/l and keep them at 50-100 μg/l during the maintenance phase.54 The Dutch guidelines differ slightly; during the maintenance phase it is sufficient to keep ferritin below the upper limit of the reference range (300 and 200 μg/l for males and females, respectively).45
With regard to genetic counselling, it is especially important to screen brothers and sisters of the patient for the presence of TFR2 mutations. Since mutations are rare, the chance that a patient’s partner is a carrier is extremely small, unless there is consanguinity. Therefore, it is normally not necessary to screen any children. The rarity as well as the variety of the mutations also implicate the absence of solid data on the genotype and phenotype relation.55 For our cases, it is at least striking that the patient with the severest mutations (nonsense and splicing rather than missense) has the earliest age of onset.
Recently, Nai et al. demonstrated the role of TFR2 in the regulation of erythropoiesis using animal studies.56 They suggest that TFR2 normally decreases the sensitivity of the EPO receptor and balances the amount of erythropoiesis with iron availability to prevent depletion of tissue iron for remaining essential metabolic functions. However, when TFR2 is lost in the erythroid progenitor cell, the balance is lost as well, leading to a constant high level of erythropoietic activity. This is supported by mice without TFR2 in the bone marrow, who fail to develop anaemia in case of iron deficiency. In patient 3, extensive phlebotomy led to iron deficiency (ferritin 16 μg/l, TSAT 8.5%). Yet, the decrease in Hb was limited and she did not develop anaemia (Hb 8.2 mmol/l versus 8.5 mmol/l before phlebotomy), which would be expected considering the iron status. Hb is the product of the mean corpuscular haemoglobin (MCH) and erythrocyte count. MCH will decrease in an iron-deficient state due to haeme-regulated inhibitor kinase,57,58 a pathway that is most likely unaffected in our patient, as her MCH decreased along with the mean corpuscular volume during phlebotomy. On the other hand, as stated, loss of function of TFR2 leads to uninhibited production of erythrocytes, demonstrated by similar erythrocyte counts in the patient before and after phlebotomy. This limits the decrease in Hb and may explain why the patient did not become anaemic despite very low iron stores. We thus speculate that the loss of TFR2 regulation in the erythroid progenitor cell contributes to the lack of anaemia in this patient. Interestingly, however, both patient 1 and 2a developed mild anaemia, despite adequate iron stores as reflected by their ferritin levels.
In conclusion, early recognition of type 3 HH patients is essential to prevent irreversible and incapacitating complications. When a patient with hyperferritinaemia presents, we recommend to rule out secondary iron overload and other causes of hyperferritinaemia using Hb and TSAT. Normal Hb combined with elevated TSAT at presentation is consistent with HH and an indication for screening for the HFE p.Cys282Tyr and p.His63Asp mutations. If the common mutations in HFE are absent, a hepcidin/ferritin ratio below the lower limit of the reference range may indicate a more severe form of HH. A liver iron content > 100 μmol/g dry weight may further point towards the diagnosis of these rare forms. WES or other massive parallel sequencing techniques can then be used to screen at least HFE (whole gene), HJV, HAMP, TFR2, and SLC40A1. In cases in which causative mutations are not identified, we recommend referring the patient to a centre with expertise in rare iron disorders.
ACKNOWLEDGEMENTS
We thank Edwin van Kaauwen and Bert van den Heuvel for their help and support in DNA analysis.
DISCLOSURES
We declare that the authors have no conflict of interest.
REFERENCES