KEYWORDS
Anaemia, cardiovascular, chronic kidney disease, dialysis, hepcidin, iron
INTRODUCTION
Hepcidin was first identified in 2001 as a peptide with antimicrobial properties.1,2 It was postulated that hepcidin contributes in the defence against extracellular infection by reducing serum iron levels, as iron availability is necessary for bacterial growth and enhances oxidative stress.1 Furthermore, suppressed hepcidin expression was shown to be related to iron overload disorders, such as haemochromatosis.3 Subsequently, hepcidin was established as a key regulator of iron homeostasis and involved in the pathogenesis of anaemia of chronic disease.4,5 Anaemia is also a well-known complication in patients with chronic kidney disease (CKD). In addition to ‘true’ iron deficiency, many CKD patients have functional iron deficiency, which is characterised by impaired iron release from body stores that are unable to meet the demand for erythropoiesis.6 Availability of ‘functional’ iron is important to obtain increased haemoglobin levels by treatment with erythropoiesis-stimulating agents (ESA). Because of the above-mentioned sequalae in patients with CKD and end-stage renal disease (ESRD), the relevance of hepcidin for this patient group seems obvious. Indeed, many papers on various aspects of hepcidin in CKD and ESRD patients have been published and the expectations of the clinical utility of measuring hepcidin to guide the treatment of renal anaemia were high.7-9 In this review, several characteristics of hepcidin, such as its function and regulatory pathways, will be discussed, as well as the various measurement assays. Furthermore, the value of hepcidin with respect to its use in clinical decision-making for renal anaemia in CKD patients and its role as a (prognostic) biomarker is described. The present knowledge on the role of hepcidin in this respect will be presented as a descriptive review. A literature search was performed in PubMed and Google Scholar with the search terms hepcidin, chronic kidney disease, dialysis (hemodialysis, hemo(dia)filtration and peritoneal dialysis) and cardiovascular (disease). The search was limited to full-text articles published in English. Furthermore, references of the selected articles were screened.
HEPCIDIN: STRUCTURE AND KINETICS
Systemic hepcidin is mainly produced in the liver, but expression by almost all other cells and tissues has been described, such as kidney tubuli, the heart, retina, fat, the lungs and the pancreas as well as in monocytes, neutrophils and macrophages.10 Hepcidin is a hairpin-shaped molecule stabilised by four disulphide bridges.11,12 It is mainly present in its bioactive form hepcidin-25, a 25 amino acid peptide of 2.8 kD. Hepcidin-20, -22 and -24 are isoforms with no or unknown biological function.13 Under physiological conditions, these isoforms are present in the urine, but virtually absent in the blood.14,15 However, in patients with CKD, and especially in those on dialysis, both serum levels of hepcidin-25 and its isoforms are elevated.13,16-18
Hepcidin-25 can circulate freely, or it can be bound to α2 -macroglobulin and to a lesser extent to albumin.19,20 The extent to which hepcidin is protein bound is not clear and estimates of the freely circulating fraction vary from 11 to 98%.19,20 Protein-bound hepcidin might be biologically more active than unbound hepcidin, as was shown in mice.21 However, this could be explained by the fact that clearance of protein-bound hepcidin is diminished, thereby increasing its half-life and also its activity.
Clearance of hepcidin is assumed to occur via cellular degradation at its sites of action, and via excretion with the urine. In healthy individuals, the fractional excretion of hepcidin is negligible (never exceeding 3-5%),22,23 implicating that under normal conditions, hepcidin is either almost completely reabsorbed by the tubuli,24 and/or degraded in kidney tubules, and/or not freely filtered by the glomerulus (due to its protein-bound character). Furthermore, in dialysis patients, it has been shown that hepcidin levels are influenced by mutations and polymorphisms in genes encoding proteins that are involved in the expression of hepcidin. Examples of these proteins are the haemochromatosis protein (HFE) and matriptase-2 (encoded by the TMPRSS6 gene), resulting in lower and higher hepcidin levels, respectively.25,26 In the general population, the associations between polymorphisms in HFE and TMPRSS6 with hepcidin are less clear.27
HEPCIDIN: FUNCTION AND REGULATION
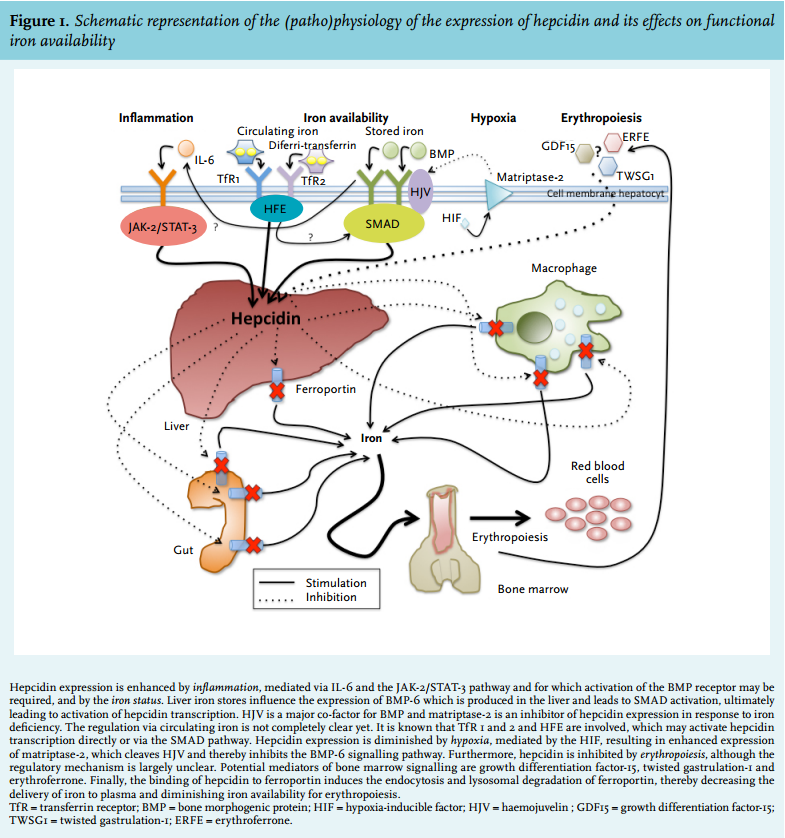
Hepcidin is able to express its regulatory function in iron homeostasis by binding to ferroportin on the membranes of iron-exporting cells, such as hepatocytes, macrophages and enterocytes (figure 1).28,29 Ferroportin is a transmembrane efflux channel that transfers cellular iron to the plasma. The binding of hepcidin to ferroportin induces endocytosis and lysosomal degradation of ferroportin, thereby decreasing the delivery of iron to plasma and diminishing iron availability for erythropoiesis.28,30 Furthermore, it has been reported that hepcidin can bind iron directly.10,31 However, the proportion of hepcidin which binds iron, its binding capacity and the relevance of this process in vivo are unknown.
Hepcidin expression can be modulated through several mechanisms (figure 1). Hepatic hepcidin production is increased by one of two main regulatory signals:
(1) Elevated body iron availability. This results in inhibition of further iron adsorption from the gastrointestinal tract. Hepcidin expression can be both regulated by stored iron and by circulating iron (reviewed by Meynard et al.) 32 but many aspects of this regulatory pathway are still not completely clear. Hepatic iron accumulation leads to increased expression of bone morphogenic protein-6 (BMP-6) in the liver. This results in nuclear translocation of SMAD proteins and subsequent activation of hepcidin transcription. Haemojuvelin (HJV) is a major co-factor for BMP and matriptase-2 is an important inhibitor of hepcidin expression in response to iron deficiency, possibly by cleaving HJV.32 Concerning hepcidin regulation by circulating iron, much is still unclear. It is likely that transferrin-bound iron is bound by transferrin receptor (TfR) 1 and 2, and that HFE is involved, which possibly activates the SMAD pathway. However, the exact links between TfR 1 and 2, HFE and the SMAD pathway are unknown.32,33
(2) Inflammation. This is mainly mediated via IL-6 and the IL-6/Janus kinase 2 (JAK2)-signal transducer and activator of transcription 3 (STAT3) pathway.30,32-34 Recent data in mice suggest that the BMP receptor, which is also involved in regulation of hepcidin by hepatic iron stores (see above), is required for induction of hepcidin expression by IL-6.35
Hepcidin production is inhibited by low circulating iron levels and low iron stores (see above) and two main other regulatory signals:
(1) Increased erythropoietic activity, such as after ESA administration. The exact underlying mechanism is largely unclear, but it is most probably mediated by molecules released by erythroid precursors, such as growth differentiation factor 15, twisted gastrulation protein homologue 1 and erythroferrone.36
(2) Hypoxia, mediated by the hypoxia-inducible factor (HIF), resulting in enhanced expression of matriptase 2 (encoded by the TMPRSS6 gene).37 This protein cleaves HJV and thereby inhibits the BMP-6 signalling pathway.33,38
HEPCIDIN: MEASUREMENT TECHNIQUES
Measurement of hepcidin in human sera has been challenged by the fact that its structure with both hydrophobic and hydrophilic regions results in adsorption to surfaces (e.g. to the plastic of the blood tubes).10 Furthermore, the creation of antibodies for immunochemical assays is complicated due to the small size of hepcidin, the compact structure caused by the disulphide bridges and its highly conserved sequence among species which impedes an immune response in host species.39 Finally, under physiological circumstances, hepcidin concentrations increase during the day.40 This circadian rhythm is explained by an innate diurnal rhythm, rather than by dietary iron intake during the day.41 In dialysis patients, this circadian rhythm has been reported to be virtually absent.42 Currently, two types of hepcidin quantification methods have been studied in patients with CKD, namely (1) immunochemical assays, including radioimmunoassays and ELISA, of which some are commercially available. Most of these techniques measure both biologically active and inactive isoforms; and (2) mass spectrometry, which is more expensive and technically demanding, but able to detect each isoform separately.10,13,15,43 Thus far, the gold standard hepcidin assay has not yet been defined. Besides, several questions remain.
First, there is a considerable inter-assay variability and measured values can vary by a factor 10.10,43-45 This precludes the definition of universal reference intervals and clinical decision limits. Nevertheless, within- and between-sample variation is fairly similar for most assays and hepcidin levels obtained are mutually correlated.44,45 However, some commercial assays do not correlate with many other assays and do not provide physiologically meaningful results; these include the prohepcidin kits.
Second, with most immunoassays, it is unclear if total hepcidin (sum of hepcidin-20, -22, -24 and -25) is measured, or whether bio-active hepcidin-25 is specifically quantified (with specific hepcidin-25 antibodies or two-site ELISA).46
Third, in general, it is not yet fully established whether protein-bound or free hepcidin is measured. Since protein-bound hepcidin has been reported to be more biologically active,21 it cannot be completely ruled out that this might be of importance.
Initiatives have been undertaken to standardise hepcidin results worldwide and to enable the definition of reference values and clinical decision limits for different patient categories, such as CKD patients.44,45 Since reliable calibrators are still lacking, algorithms have been constructed enabling different laboratories to calculate a HEPcidin CONsensus (HEPCON) value using their own hepcidin results, measured with both mass spectrometric and immunochemical assays.45 This is an important step towards harmonisation of different assays, which may facilitate the interpretation and comparison of different studies.
HEPCIDIN AND CKD
It has been well established that hepcidin levels are increased in non-dialysis CKD patients as well as in dialysis patients, possibly due to increased production (driven by inflammation and elevated stored body iron levels) or reduced clearance.9,17,47 In one study from Italy, however, hepcidin levels measured with mass spectrometry were similar in 199 haemodialysis patients and 188 age- and sex-matched controls.25 This could be explained by careful matching and inclusion of haemodialysis patients, namely those who received relatively little iron supplementation and were not iron-loaded.48 These data need confirmation.
Hepcidin and its correlation with eGFR
Research on the relation between hepcidin and the estimated glomerular filtration rate (eGFR) has shown conflicting results. In a study in CKD patients, total hepcidin (sum of all isoforms) measured with an ELISA was associated with eGFR.9 In other studies in this patient category, in which hepcidin-25 was measured with mass spectrometry, this association was not present.17,49,50 Previously, it has been suggested that the relation between eGFR and hepcidin might be due to the measurement of inactive isoforms in non-specific assays.17 However, the bioactive form of hepcidin measured with a hepcidin-25 specific radioimmunoassay was associated with eGFR.42 Furthermore, in a cohort of over 400 chronic haemodialysis patients, hepcidin-25 levels assessed by mass spectrometry were correlated with the eGFR measured by an interdialytic 24-hour urine collection in univariate and multivariate models.51 In a study in 199 non-dialysis CKD patients, it was shown that hepcidin levels increase as eGFR decreases, although those patients who were markedly iron deficient still had low levels of hepcidin at low eGFR values. These data suggest that hepcidin still reflects iron status at low eGFR levels.47 In conclusion, hepcidin levels correlate inversely with eGFR, but this relation is blunted by iron deficiency in patients with low eGFR.
Removal of hepcidin with renal replacement techniques
Hepcidin can be removed from the blood by both haemodialysis and peritoneal dialysis.16,17,52-56 Hepcidin reduction was similar after treatment with different dialysers57,58 and reduction tended to be superior with haemodiafiltration treatment.53 After a haemodialysis treatment, hepcidin was present both in the ultrafiltrate and bound to the dialyser membrane.17 However, sustained lowering of hepcidin levels with extracorporeal renal replacement therapy does not occur, since after an initial decrease in hepcidin levels, post-dialysis levels were already back to pre-dialysis values as soon as one hour after the end of a dialysis session.17,52
Correlations between hepcidin parameters of inflammation and iron status
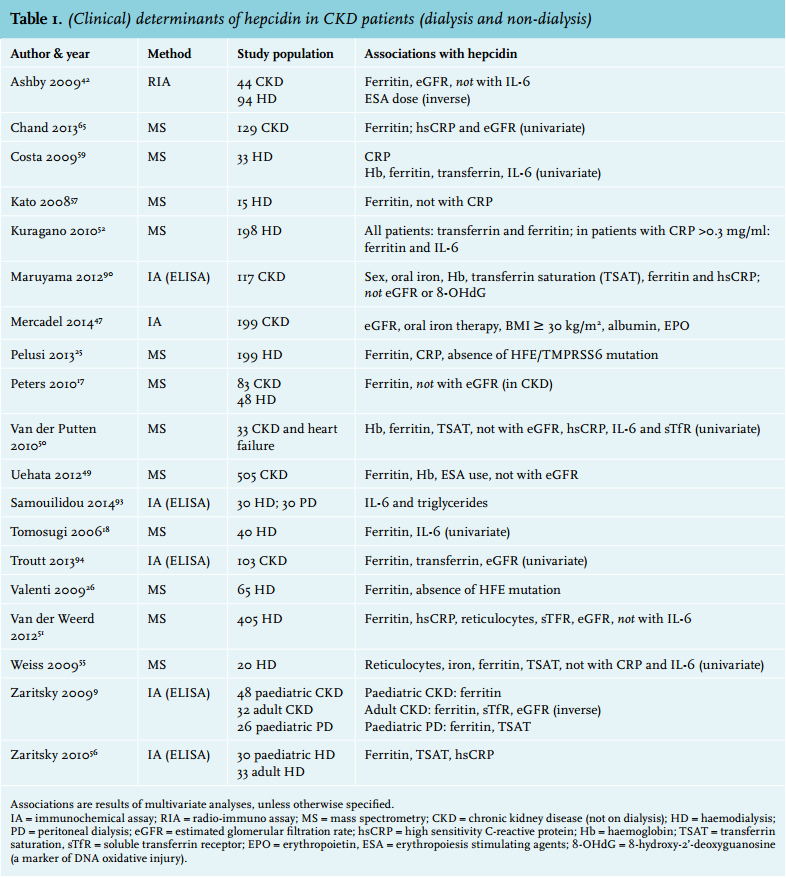
It is well known that anaemia, iron deficiency and inflammation are highly prevalent in CKD patients. As this might all be associated with (or explained by) elevated hepcidin levels, many studies in CKD patients correlating hepcidin with various clinical parameters have been performed (table 1). Virtually all studies on hepcidin, including those in dialysis patients, have observed a strong association between ferritin and hepcidin.17,51,52,55-57 Concerning markers of inflammation, several studies have shown a relation with C-reactive protein (CRP)17,51,56,59,60 or IL-651,59 in small groups of chronic haemodialysis patients, whereas others did not.55,61 In a study in over 400 chronic haemodialysis patients, it was demonstrated that the relation between hepcidin-25 and ferritin was present in all strata of inflammation (as measured by high-sensitivity CRP), whereas the relation between hepcidin-25 and high-sensitivity CRP was only present when ferritin was not markedly elevated (< 530 ng/ml; figure 2).51 This finding suggests a stronger relation between hepcidin-25 and ferritin than between hepcidin-25 and specific markers of inflammation.
HEPCIDIN: A TOOL FOR CLINICAL DECISION - MAKING IN RENAL ANAEMIA?
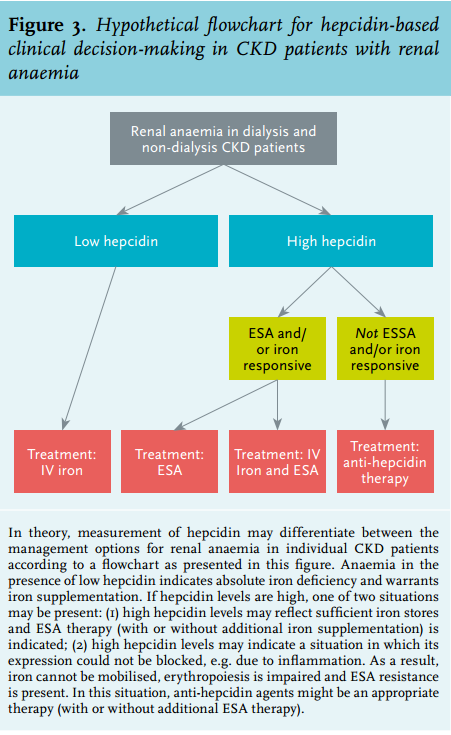
Based on early insights into hepcidin regulation and function, it has been speculated that measurement of hepcidin could be applied in clinical assessment of anaemia in CKD patients, including those on dialysis.62 In CKD patients, anaemia in the presence of low hepcidin indicates absolute iron deficiency and warrants iron supplementation. In contrast, in anaemia with high hepcidin levels, one of two situations may be present: (1) high hepcidin levels may reflect sufficient iron stores and ESA therapy (with or without additional maintenance iron supplementation) is indicated; (2) high hepcidin levels may indicate a situation in which its expression could not be inhibited, e.g. due to inflammation. As a result, iron cannot be mobilised (i.e. ‘functional iron deficiency’), erythropoiesis is impaired and ESA resistance is present. In this situation, ESA or iron supplementation will not result in an increase in haemoglobin levels and anti-hepcidin agents (with or without additional ESA therapy) might provide an appropriate therapy. However, anti-hepcidin agents are not yet approved for clinical use. In conclusion, measurement of hepcidin may theoretically differentiate between the management options for renal anaemia in individual CKD patients according to a flowchart as presented in figure 3.
Hepcidin and ESA therapy
It has been suggested that hepcidin might contribute to ESA resistance,8 since animal data have shown that overexpression of hepcidin impaired the response to even very high ESA doses.63 In humans, however, data on the relation between hepcidin levels, ESA treatment and ESA resistance are inconsistent. In haemodialysis patients and in patients with the cardio-renal syndrome, hepcidin levels decreased after ESA administration.50,55,64 In cross-sectional data of almost 100 haemodialysis patients on ESA maintenance therapy, hepcidin levels were lower in patients receiving higher ESA doses, regardless of the haemoglobin level that was reached.42 However, in another cross-sectional study in over 400 haemodialysis patients, hepcidin levels were not associated with either the dose of ESA or intravenous iron.51 Furthermore, two small studies revealed that hepcidin-25 levels were similar in ESA-responsive and ESA-resistant haemodialysis patients, suggesting that the assessment of hepcidin is not helpful in deciding which individual patient may benefit from ESA therapy.57,59 Nevertheless, in patients with the cardio-renal syndrome, ESA responders showed higher hepcidin-25 levels than non-responders, suggesting that hepcidin might rather be a marker of ESA responsiveness than associated with ESA resistance.50 Of note, in all the above-mentioned studies, bioactive hepcidin was measured (either with mass spectrometry or with a specific radioimmunoassay).
Hepcidin and iron therapy
Concerning the utility of hepcidin in assessing iron stores and managing iron supplementation therapy, overall, negative results have been reported. In a study in 56 chronic haemodialysis patients, neither hepcidin-20 nor hepcidin-25 could predict an increase in haemoglobin levels after administration of intravenous iron.61 Notably, in this study, ROC curve analysis showed that also ferritin failed to accurately predict a response, whereas the percentage of hypochromic red blood cells was the only biomarker independently associated with iron responsiveness. Furthermore, in a small group of haemodialysis patients, hepcidin-25 levels were similar before and after the administration of intravenous iron.55 In another study in 129 consecutive non-dialysis CKD patients, hepcidin-25 was able to predict a haemoglobin response after intravenous iron administration.65 However, ferritin and transferrin saturation had similar predictive utility. Again, in all the mentioned studies, hepcidin was measured with mass spectrometry.
Limitations of available evidence
In conclusion, thus far, conflicting and even forthright negative data are available to answer the question whether measurement of hepcidin may be a suitable and unique tool for clinical decision-making in ESA and iron management in CKD patients. In assessing iron stores, hepcidin did not appear to be superior over ferritin.66 An important aspect further hampering the use of hepcidin as a management tool is that in stable haemodialysis patients, intra-individual hepcidin levels, measured with both ELISA and mass spectrometry, varied widely in a relative short period of time, likely dependent on fluctuations in the inflammatory state.60,67 These studies implicate that short-term measurements of serum hepcidin in individual (dialysis) patients might not be appropriate to guide clinical decisions regarding ESA or iron management.
Moreover, at this point it should be noted that comparing the available studies is complex and hazardous as a result of the large variety in experimental circumstances. Available studies differ with respect to patients’ iron stores, levels of inflammation, stages of renal failure, dialysis regimens, doses of iron supplementation and ESA. Besides, studies use different hepcidin assays and there is a variable lag time between ESA and iron administration and blood sampling. Furthermore, most available studies included a limited number of patients precluding multivariate statistics.68
The above-mentioned aspects make systematic reviews on the usefulness of hepcidin measurements in anaemia management virtually impossible. Therefore, prospective clinical studies investigating the utility of a hypothetical treatment algorithm as shown in figure 3 are warranted.
HEPCIDIN: A BIOMARKER FOR CARDIOVASCULAR DISEASE?
In a study in 335 non-dialysis CKD patients, it was shown that hepcidin-25, measured with mass spectrometry, predicted the progression of renal anaemia both in iron replete and deplete patients, during a median follow-up period of 3.6 years.69 This observation demonstrates the concept of hepcidin as a biomarker, in this specific case for renal anaemia. Recent evidence also shows a new conceptual role for hepcidin as a biomarker for cardiovascular disease. The involvement of hepcidin in the development of atherosclerosis and cardiovascular disease is supported by several (pre-)clinical studies, which will be briefly reported below.
The role of hepcidin in atherosclerotic disease
Several experimental animal studies provided evidence for the involvement of hepcidin in atherosclerotic processes. In mice, suppression of hepcidin resulted in reduced intracellular iron content in macrophages, resulting in an augmented efflux capacity of cholesterol.70 Furthermore, these animals exhibited diminished foam cell formation and less atherosclerosis. In an atherosclerotic mice model, hepcidin promoted plaque destabilisation by inducing inflammatory cytokine release, intracellular lipid accumulation, oxidative stress and apoptosis of macrophages with iron retention.71 In an experiment in humans, intracellular iron content in monocytes derived from atherosclerotic plaques was increased in the presence of hepcidin, which resulted in enhanced reactive oxygen substances preventing cholesterol efflux from these cells.72 In clinical and epidemiological studies, associations have been found between hepcidin and markers of vascular stiffness, atherosclerosis and cardiovascular disease. In 143 patients with non-alcoholic fatty liver disease, hepcidin-25 levels were associated with the presence of carotid plaques.73 In a similar group of 130 patients with non-alcoholic fatty liver disease, monocyte chemo-attractant protein-1, a chemokine that plays a crucial role in both the initiation and progression of atherosclerosis, was correlated with hepcidin-25 and an independent predictor of the presence of atherosclerotic plaques.74 In 60 patients with rheumatoid arthritis, hepcidin was correlated with coronary artery atherosclerosis, measured by a coronary calcium score, even after multivariate adjustment.75 Finally, in a Dutch population-based cohort of 766 post-menopausal women, hepcidin was associated with the presence of plaques in the carotid artery, adjusted for eGFR, inflammation and traditional cardiovascular risk factors.76
Recently, the role of hepcidin as a cardiovascular marker gained interest in the high-risk CKD population.77 Two studies, one in 168 chronic haemodialysis patients and another in 56 patients on peritoneal dialysis, showed an association between arterial stiffness (measured with brachial-ankle pulse wave velocity and flow-mediated dilatation, respectively) and hepcidin.78,79 Furthermore, two studies investigated the relation between hepcidin and left ventricular mass index (LVMi). One study in 146 CKD patients not on dialysis showed that lower hepcidin levels were associated with higher LVMi, possibly due to the concomitant iron deficiency resulting in an anaemic state.80 In a study in 327 chronic haemodialysis patients, no association between hepcidin and LVMi was observed.81 Finally, in a cohort of 405 chronic haemodialysis patients with a median follow-up of three years, hepcidin-25 levels were associated with the incidence of cardiovascular events, even after stepwise adjustments of clinical and anaemia-related parameters, including inflammation.82 Of note, the association between hepcidin-25 and all-cause mortality was attenuated after adjustment for inflammatory markers. Although these associations certainly do not prove causality, this study adds evidence to the hypothesis that hepcidin may be directly or indirectly involved in the pathogenesis of cardiovascular disease and mortality in patients on dialysis and therefore may function as a biomarker.
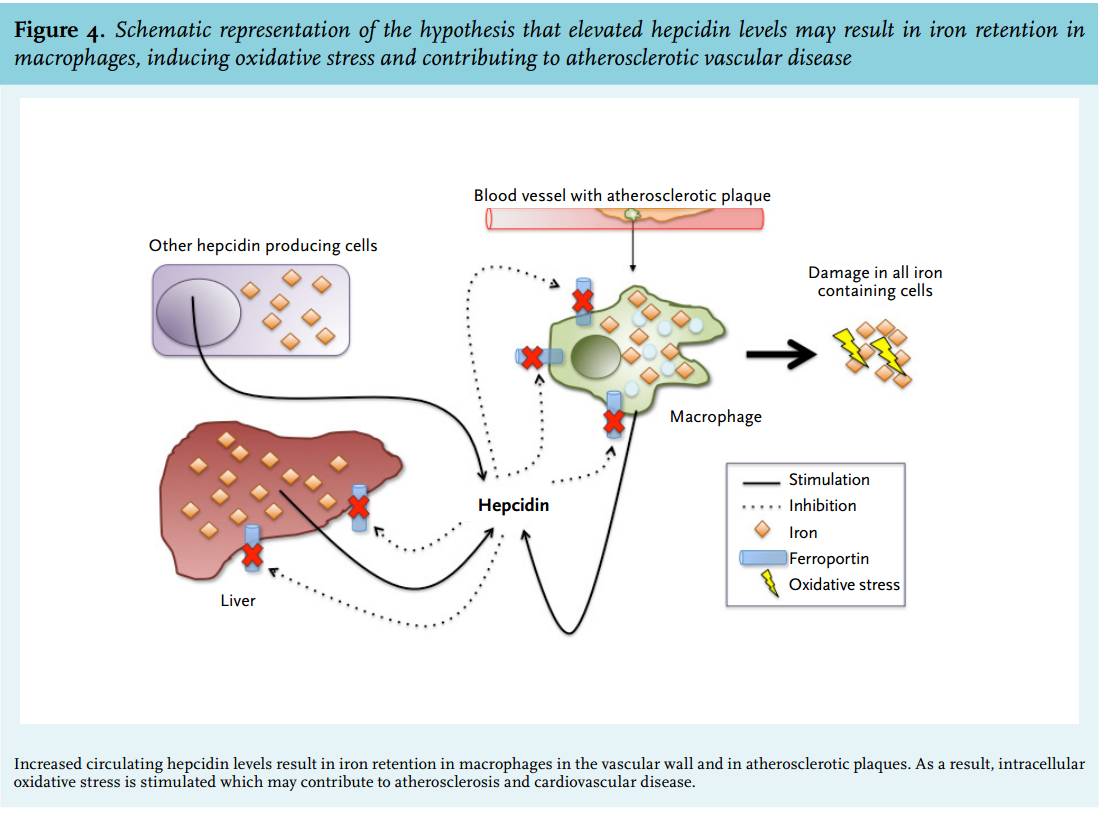 Hepcidin and atherosclerotic disease: pathophysiological concept
Hepcidin and atherosclerotic disease: pathophysiological concept
As mentioned before, hepcidin is predominantly synthesised in the liver, but low levels of expression in other cells, including macrophages, are present, enabling local fine-tuning of systemic iron regulation.38,83 Vice versa, systemic hepcidin, mainly produced in the liver, can exhibit an inhibitory effect on iron release from macrophages.7,10 It can be hypothesised that elevated hepcidin, either produced locally or systemically, is associated with atherosclerotic disease by retaining iron in macrophages in the vascular wall. This intracellular iron sequestration may result in a pro-atherogenic environment, possibly mediated by oxidative stress, inflammatory responses and macrophage apoptosis, and resulting in clinical events, such as myocardial ischaemia, stroke and peripheral vascular disease (figure 4).71,84,85
In a study in an atherosclerotic mice model, however, this ‘iron hypothesis’ of cardiovascular disease could not be confirmed.86 These animals exhibited iron accumulation specifically in macrophages due to an additional ferroportin mutation, or they were iron loaded due to parenteral administration of high iron doses. In these mice, hepatic hepcidin expression was not enhanced and atherosclerotic plaque size was not increased. The authors explain their findings by suggesting that in macrophages, antioxidant defence strategies may be very efficient.86
In CKD patients, several additional remarks with regard to the ‘iron hypothesis’ of cardiovascular disease need to be considered. First, since the cardiovascular risk profile of CKD patients seems to differ from the general population,87 the question arises whether the atherogenic effect of iron accumulation and oxidative stress in the vascular wall, as has been observed in non-CKD patients, is relevant in this specific high-risk CKD patient group as well. Second, it is not known whether iron sequestration in macrophages plays the same role in atherosclerotic plaques in the arterial intima as in calcified plaques in the arterial media, as the latter is a characteristic of cardiovascular disease in dialysis patients.88,89 Finally, the relation between hepcidin and oxidative stress in CKD patients has not yet been unequivocally demonstrated.90 Nevertheless, the available experimental and epidemiological evidence of the complex interplay between hepcidin, iron accumulation in macrophages in the vascular wall and cardiovascular disease is hypothesis generating for further research on the potential usefulness for hepcidin as a cardiovascular biomarker, especially in the vulnerable CKD population.
CONCLUSIONS AND REMAINING QUESTIONS
In this review, currently available pre-clinical and clinical data have been summarised in order to weigh the merits of hepcidin in three areas of interest.
1. Hepcidin and the pathophysiology of renal anaemia in CKD patients.
The identification of hepcidin provided relevant information regarding the understanding of the concept of functional iron deficiency in dialysis and non-dialysis CKD patients, and it revealed a possible mechanism for the presence of ESA resistance despite extensive intravenous iron loading. In addition, knowledge on the clinical characteristics of hepcidin, such as its inflammation-driven regulatory pathway and its renal clearance profile, made it a relevant peptide in CKD patients. However, several features restrict its clinical use at the moment: pre-analytical handling of hepcidin is critical, the accessibility of the most accurate and precise assay for hepcidin-25 is limited and the clinical relevance of specifically measuring hepcidin-25 instead of total hepcidin by more accessible ELISA assays is unclear. Available studies are difficult to compare since absolute levels obtained by various assays that are used worldwide differ substantially. This precludes the assessment of universal clinical decision limits. Furthermore, other important characteristics of hepcidin, such as its protein binding, have not yet been completely elucidated.
2. Hepcidin as a diagnostic tool and for guiding clinical decisions in CKD patients.
Hepcidin levels in dialysis and non-dialysis CKD patients are elevated, due to decreased (renal) clearance and/or enhanced production, e.g. by inflammation and iron loading. Its levels are highly correlated with ferritin in virtually all studies, and therefore the question arises what the advantage is of measuring hepcidin as compared with measuring ferritin, which is easier and less expensive. Inflammation is an important factor in the expression of hepcidin and most studies show an association between hepcidin and inflammatory markers such as CRP and IL-6. Studies on the role of hepcidin in distinguishing between a situation of ESA responsiveness or ESA resistance have shown negative or inconsistent results. Similarly, iron responsiveness could not be predicted by hepcidin levels. Furthermore, the high intra-patient variability makes hepcidin unsuitable as an instrument to guide treatment in individual patients. Therefore, it can be concluded that currently, hepcidin cannot be considered a valuable clinical tool in diagnosing and treating renal anaemia.
3. Hepcidin as a prognostic biomarker for cardiovascular disease.
Several experimental, clinical and epidemiological studies in animals and humans showed an association between hepcidin and atherosclerotic disease and clinical events, possibly mediated via iron sequestration in macrophages in the vascular wall. These observations point towards a role for hepcidin as a biomarker for cardiovascular disease, which is also shown in several clinical reports in dialysis patients.
Future perspectives
Finally, although beyond the scope of this review, the presence of numerous anti-hepcidin compound initiatives that are currently under way and use hepcidin as a target of therapy, should be acknowledged.6,34,91 Although no clinical studies of anti-hepcidin therapy in patients with CKD have been published yet, the first results of an anti-hepcidin antibody tested in humans are promising: administration of this antibody in humans was able to block inflammation-induced reduction in serum iron.92 Whether, in the future, measuring hepcidin could be useful to establish an indication for anti-hepcidin therapy, or whether it could be helpful in monitoring anti-hepcidin therapy, needs to be determined.
Taken together, hepcidin has contributed to our understanding of the pathophysiology of iron misdistribution and anaemia in CKD, but it has not fulfilled its promise as a diagnostic and management tool thus far. Its role as a biomarker of cardiovascular disease is promising and needs confirmation.
DISCLOSURES
D.W.S. is a co-founder and medical director of the ‘Hepcidinanalysis.com’ initiative (www.hepcidinanalysis. com). The other authors declare no conflicts of interest.
REFERENCES