KEYWORDS
SIADH, syndrome of inappropriate antidiuretic hormone, traumatic brain injury, TBI
BACKGROUND
The syndrome of inappropriate antidiuretic hormone secretion (SIADH) causes excessive antidiuretic hormone (ADH) release, leading to water retention and, as a consequence, hyponatremia. Laboratory results show hyponatremia < 135 mmol/l, together with decreased effective serum osmolality < 275 mOsm/kg.1 Typically, the urine sodium concentration is above 40 mmol/l, with a concomitant high urine osmolality > 100 mOsmol/kg. The patient is clinically euvolemic. Depending on the severity of the hyponatraemia, symptoms can be subtle or severe, comprising nausea, lethargy, an altered mental status, convulsions, and coma.
SIADH has been linked to a variety of aetiologies and is most commonly seen in combination with (pulmonary) malignancy, surgery, drugs, and all types of central nervous system (CNS) disruptions. One of these CNS disruptions is traumatic brain injury (TBI). The exact pathophysiology of SIADH after TBI has not yet been clarified. Most hypotheses encompass either damage to the pituitary stalk or the posterior pituitary, leading to an inappropriate secretion of ADH.2,3 Risk factors for the development of SIADH after TBI have not yet been elucidated.4 There is no unambiguous indication to assume that the severity of the trauma is correlated with the development of pituitary dysfunction.2,4 The objective of this case report is to draw attention to the sometimes long-lasting nature that SIADH after TBI can display. Literature on the emergence of SIADH shortly after TBI is available, but only one case of ongoing SIADH (six years in our case) has been previously described.3
CASE PRESENTATION
In February 2013, a 59-year-old woman suffered a traumatic brain injury (TBI) after falling down the stairs. She suffered a bilateral mastoid fracture, which led to paralysis of the facial nerve and a central scull base fracture. She was admitted to the hospital for one week for supportive care. At admission, her serum sodium level was 141 mmol/l (reference range: 135-145 mmol/l). Two weeks after the fall, she was admitted to the inpatient clinic again for elective facial nerve decompression. Upon admission, she complained of nausea, vomiting, tiredness, and dizziness. Her blood pressure was 138/92 mmHg with a heart rate of 84 beats per minute, and she had normal urine production, serum creatinine, and blood urea nitrogen (table 1). Laboratory results revealed hyponatraemia with a serum sodium concentration of 114 mmol/l. The results seemed to be consistent with SIADH, showing a low serum osmolality and a high urine sodium and urine osmolality (figure 1, day 0). 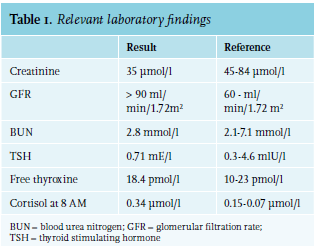
There were no signs of hyperthyroidism or adrenal insufficiency (table 1), and secondary adrenal insufficiency was determined improbable by a normal metyrapone test. The patient did not use any relevant medication and there were no signs of malignancy, disease of the central nervous system, or pulmonary disease. Furthermore, a CT scan of the cerebrum did not show abnormalities in the hypothalamus/pituitary region or olfactory neuroblastoma. An MRI revealed an absence of the normal hyperintense signal of the posterior hypophysis, which has been correlated with various forms of SIADH in small series (figure 2).5 Therefore, it was concluded that SIADH caused by TBI was the most likely explanation. 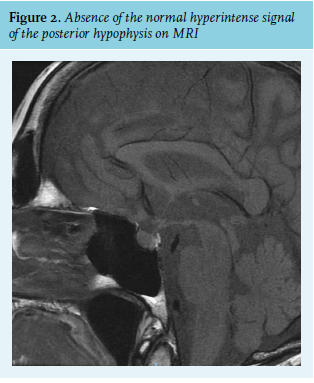
Initially, the patient was treated with a fluid restriction of 1 l/24 h and sodium 3% infusions, leading to the normalisation of sodium levels (135 mmol/l) and fading of symptoms. On day 15, the patient was discharged with a fluid restriction of 1 l/24 h and oral sodium chloride tablets (see figure 1 for details about the dosage and laboratory results following this regimen). Details about the laboratory results in relation to adjustments in therapy and water load tests are summarised in table 2* and table 3*, respectively.
Over the years, the therapy was adjusted to a fluid restriction of 1.6 l, there was an increase in the sodium tablet dosage, and urea powder was added. The patient was able to adhere well to this regimen. In the following years, we planned several water load tests to test whether the SIADH was still profound (figure 1 at 4 months, 14 months, and after 5 years). The first two tests failed, as the serum sodium level decreased as soon as the patient quit taking her sodium and urea supplementation and increased her water intake. Five and a half years after her fall, we observed a spontaneous increase of sodium levels (142 mmol/l) whilst the patient was still on a fluid restriction regimen and taking sodium chloride and urea powder. A third water load test demonstrated that the ability to dilute the urine had been restored, indicating appropriate ADH secretion. In the following months, despite unrestricted fluid intake, the sodium measurements remained normal, and we could declare the SIADH as cured.
DISCUSSION
In this report, we demonstrate a case of long-lasting SIADH after traumatic brain injury, which eventually resolved spontaneously after five years. Several cases of SIADH after TBI have been described, but to our knowledge, only one other case has illustrated SIADH withstanding over several years.3
Born et al. described 109 patients with a Glasgow Coma Scale < 7 after a severe head injury. Thirty-six patients developed SIADH, all within three weeks after the head injury had occurred. No long-term follow up of the sodium levels was described, but it was assumed that, as is in most cases, SIADH resulting from TBI is of a transient nature.2 Chen et al. described four cases of SIADH after TBI, for which SIADH developed within four days after TBI.6 These patients all received supportive care consisting of fluid restriction, diuretics and, in some cases, saline supplementation; in all cases, the SIADH resolved within 10 days.
To the best of our knowledge, only one case of persistent SIADH has been published. Dick et al. described the case of a 32-year-old man who developed SIADH after TBI caused by a high-speed motorcycle accident.3 The patient was treated with a fluid restriction of 1.2 l/day. The patient had difficulties adhering to the fluid restriction. He was therefore readmitted several times over the following four years, showing recurrences of hyponatraemia consistent with SIADH. Eventually, demeclocycline was started, which kept serum sodium levels in the normal range, even after fluid restriction was stopped. The authors concluded that the resolution of hyponatraemia was due to treatment with demeclocycline and is unlikely to reflect a spontaneous resolution of the SIADH. However, with our case, we have shown that spontaneous recuperation after more than five years is possible.
Given the observation that SIADH can persist over several years, treatment should be adjusted for the individual patient, especially since adherence to treatment can be difficult. The cornerstone of treatment is fluid restriction. In cases of acute and/or symptomatic hyponatraemia, treatment with hypertonic saline is recommended. Urea powder can increase serum sodium levels by increasing urinary osmolality and water clearance, and can, if necessary, be combined with loop diuretics. The addition of oral salt tablets can increase the serum concentration of sodium. In the setting of persisting hyponatraemia caused by SIADH, the use of salt and urea tablets can somewhat relax the fluid restriction.3 In addition, vasopressinantagonists (e.g., tolvaptan) can be used to increase the serum sodium levels by inducing free water clearance in the distal parts of the nephron.7 A similar effect can be obtained with the use of demeclocycline, which diminishes the responsiveness of the collecting duct to ADH.2 With our patient, this was not indicated because of her compliance to the fluid restriction and oral salt supplementation and urea powder.
CONCLUSION
Having reviewed the current literature, we found that, besides the case reported by Dick et al., no other studies have been published on SIADH lasting several years after the initial TBI. We have demonstrated a case of persistent SIADH after TBI, which resolved spontaneously after over five years of treatment.
DISCLOSURES
All authors declare no conflicts of interest. No funding or financial support was received.
*Tables 2 and 3 are available from the authors upon request.
REFERENCES