KEYWORDS 
Acute kidney injury; enteric hyperoxaluria; fat malabsorption; nephrolithiasis; oxalate nephropathy; oxalosis; secondary oxalosis
INTRODUCTION
Oxalate nephropathy is a rare cause of renal insufficiency. It is characterised by damage to the interstitium and tubuli of the kidneys, caused by deposition of oxalate crystals. During the past decade, oxalate nephropathy has been described several times as a complication of fat malabsorption after bariatric surgery, and in patients with short bowel syndrome, cystic fibrosis, celiac disease, and exocrine pancreatic insufficiency. Today, the diagnosis is not very rare; for example, one study identified that in a series of 611 patients referred for pre-transplantation consultation, 17 (3%) had oxalate nephropathy.1
We describe two patients with exocrine pancreatic insufficiency, who were evaluated in our hospital because of acute renal insufficiency of unknown origin.
Patient A
A 77-year-old male was referred to our centre with deterioration of renal function. He had a history of hypertension, hypercholesterolaemia, and a pT3N1 pancreatic head carcinoma, which had been treated by pancreaticoduodenectomy and adjuvant chemotherapy two years earlier.
There was no history of kidney stones and the family history was negative for renal diseases. Six months after the pancreaticoduodenectomy, the patient started taking pancreatic enzyme supplements (pancreatin) because of chronic diarrhoea. After surgery, his serum creatinine level was 90 µmol/l, but increased to 180 µmol/l two months later, after a brief period of heart failure due to gemcitabine. His renal function remained stable for the following five months; however, shortly before being referred to our centre, his serum creatinine level suddenly increased to over 300 µmol/l.
The patient’s history revealed no clues for a prerenal cause of his renal insufficiency: the patient reported no vomiting, used no new medications or supplements, and had not used any non-steroidal anti-inflammatory drug. His diarrhoea ceased when he started taking pancreatin. There were no signs of a systemic disease and physical e xamination showed no abnormalities: his blood pressure was 138/75 mmHg with a heart rate of 60 beats per minute.
At the initial visit in our centre, his serum creatinine level had increased to 326 µmol/l (table 1). Further investigation showed normal anion gap metabolic acidosis (pH 7.22), normocytic anaemia (Hb 6.3 mmol/l), and hyperphosphataemia (serum phosphorus 1.76 mmol/l). Urinary examination showed some leukocytes, without erythrocytes. Twenty-four-hour urinalysis revealed a proteinuria of 0.25 grams/day. Abdominal ultrasound showed normal kidneys, measuring 10.6 and 10.1 cm. There were no signs of nephrocalcinosis. The differential diagnosis was tubulointerstitial nephritis, cholesterolemboli, or acute tubular necrosis. A kidney biopsy was performed.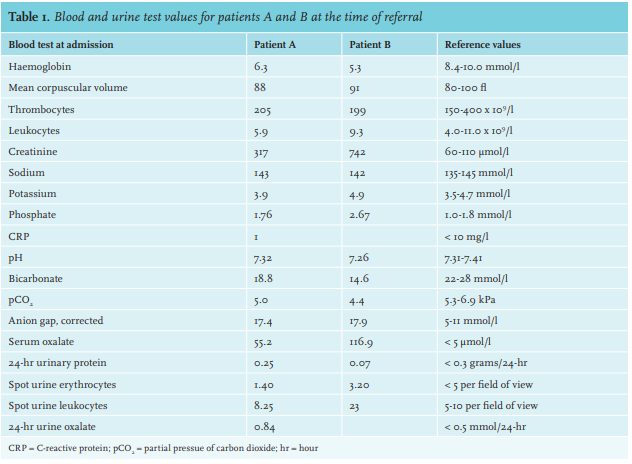
 The biopsy showed pronounced tubulopathy with extensive deposits of birefringent oxalate crystals (figure 1). The glomeruli were normal and immunofluorescence revealed no immune depositions. Thus, the patient was diagnosed with secondary hyperoxaluria due to malabsorption following exocrine pancreatic insufficiency. His serum oxalate level was 55.3 µmol/l (reference value < 5 µmol/l). Considering his age and medical history of pancreaticoduodenectomy, a primary hyperoxaluria was deemed unlikely. No mutational analysis was performed. The patient was advised to reduce dietary oxalate intake and was started on calcium supplementation before meals, together with cholestyramine. Six months later, serum oxalate levels had decreased to 30 µmol/l. Kidney function partially recovered with serum creatinine levels dropping to2 44 μmol/l (estimated glomerular filtration rate (eGFR) of 21 ml/min/1.73 m2).
The biopsy showed pronounced tubulopathy with extensive deposits of birefringent oxalate crystals (figure 1). The glomeruli were normal and immunofluorescence revealed no immune depositions. Thus, the patient was diagnosed with secondary hyperoxaluria due to malabsorption following exocrine pancreatic insufficiency. His serum oxalate level was 55.3 µmol/l (reference value < 5 µmol/l). Considering his age and medical history of pancreaticoduodenectomy, a primary hyperoxaluria was deemed unlikely. No mutational analysis was performed. The patient was advised to reduce dietary oxalate intake and was started on calcium supplementation before meals, together with cholestyramine. Six months later, serum oxalate levels had decreased to 30 µmol/l. Kidney function partially recovered with serum creatinine levels dropping to2 44 μmol/l (estimated glomerular filtration rate (eGFR) of 21 ml/min/1.73 m2).
Patient B
A 64-year-old male with a history of diabetes mellitus type 2, depression, and an exocrine pancreatic insufficiency due to chronic alcohol abuse, was referred to our hospital for dialysis following acute on progressive renal insufficiency. Due to complaints of chronic diarrhoea, he had been prescribed pancreatin by the referring physician. No further investigations into his pancreatic insufficiency had been performed, but an earlier computed tomography scan of abdomen showed extensive calcifications and atrophy of the pancreas. The patient also used thiamine, ranitidine (H2 receptor antagonist), macrogol, furosemide (loop diuretic), alfacalcidol (1-α-OH-vitamin D3), iron, and insulin.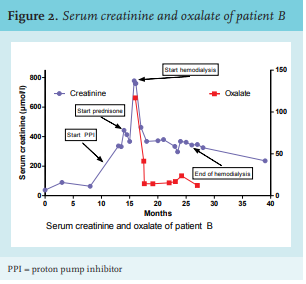
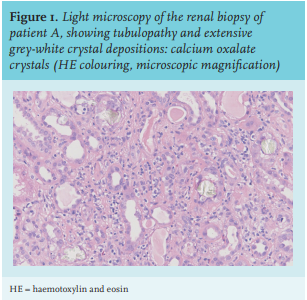
A proton-pump-inhibitor (PPI) was prescribed for reflux disease (figure 2). Upon his next visit, serum creatinine level had increased from 50 µmol/l to 222 µmol/l. Due to the suspicion of a medication-induced tubulointerstitial nephritis, the PPI was promptly ceased, and the patient was treated with oral prednisolone, 40 mg daily, without improvement of renal function.
When the patient was referred to our hospital for biopsy and dialysis, serum creatinine levels had increased to 742 µmol/l. His medical history and physical examination provided no further clues, except for signs of malnutrition. Family history was negative for kidney diseases and the patient had never suffered from kidney stones; his blood pressure was 130/84 mmHg.
The patient had normocytic anaemia (Hb 5.3 mmol/l), hyperphosphataemia (2.67 mmol/l), and metabolic acidosis (pH 7.26) (table 1). Serologic evaluation was negative, urinalysis revealed leukocyturia (23 leukocytes per field of view) with mild erythrocyturia. No crystals or casts were seen. Renal ultrasound showed kidneys measuring 12.9 and 11.6 cm, without hydronephrosis or nephrocalcinosis. The differential diagnosis consisted of IgG4 related disease, tubulo-interstitial disease, or oxalate nephropathy. IgG subclass quantification was normal.
A renal biopsy revealed tubulopathy and widespread oxalate crystal depositions, confirming the diagnosis of oxalate nephropathy. The serum oxalate was severely increased: 116.9 µmol/l (reference value < 5 µmol/l). No mutational analysis was performed.
The patient started haemodialysis and was advised to adhere to an oxalate-restricted diet. In addition, the patient took calcium supplementation and cholestyramine. After 10 months of therapy, his serum oxalate levels were stable at 14 µmol/l and haemodialysis could be ceased. Renal function eventually recovered to an eGFR 24 ml/ min/1.73 m2 (serum creatinine level 236 µmol/l) one year after ceasing haemodialysis (figure 2).
CLINICAL LESSON
Biochemistry Oxalic acid (C2H2O4) is an acid with a molecular mass of 88 kDa.2 Oxalate(C2O2-4) is the ion. It is ingested through several sources of food, notably vegetables, fruits, nuts, and grain and absorbed in the intestines, primarily in the colon. Oxalate in humans is also produced endogenously in the liver from its precursor, glyoxylate.3 Oxalate has no known biological functions.
Pathophysiology
Two forms of hyperoxaluria can be distinguished. Primary hyperoxaluria is caused by genetic defects in the hepatic degradation of glyoxalate,4 while secondary hyperoxaluria is caused by an increased intake of oxalate precursors such as ascorbic acid (vitamin C) or ethylene glycol (the main constituent of de-icer spray),5 or by an increased absorption of dietary oxalate.
In healthy individuals, calcium and oxalate form calcium oxalate, which is unavailable for enteric absorption and excreted in the stool. However, in case of malabsorption, calcium will not precipitate with oxalate but bind instead with the increased levels of intraluminal free fatty acids. The unbound oxalate is then freely absorbed into the blood, mainly through passive paracellular colonic transport.6
Malabsorption of free fatty acids and the associated steatorrhoea are seen after Roux-en-Y bypass surgery, after pancreaticoduodenectomy, and in the setting of exocrine pancreatic insufficiency. In a study with 48 patients with chronic pancreatitis, 23% had hyperoxaluria.7 Secondary hyperoxaluria is not described in patients after gastric sleeve surgery, since this does not cause steatorrhoea or malabsorption.8
Furthermore, in inflammatory bowel diseases such as morbus Crohn and celiac disease, oxalate absorption is increased due to mucosal inflammation. Elevated levels of bile acids in the intestine also seem to facilitate oxalate absorption through increased permeability.
A final cause for increased oxalate absorption is a disruption of the intestinal microbiome. Certain bacteria, such as the anaerobic Oxalobacter formigenes, degrade oxalate. (Oral) treatment with antibiotics affects these bacteria and changes the amounts of oxalate excreted in the stool.9
Oxalate in the serum is mostly filtered by the glomerulus. Research has shown that urinary oxalate excretion increases when dietary oxalate intake increases.10 Oxalate transporters are present in the proximal tubules of the kidney and in the gut. There is evidence for a bidirectional transport, so oxalate may be actively secreted and reabsorbed.11 Hyperoxaluria and the resulting calciumoxalate supersaturation in the kidney can lead to formation of calcium oxalate kidney stones, nephrocalcinosis and depositions of oxalate crystals in the renal interstitium. This process is accelerated in an acidic environment, for example, in conditions such as metabolic acidosis. Dehydration promotes crystal formation due to more concentrated urine. Secretion of ADH (anti-diuretic hormone) and activation of the renin-angiotensin-aldosterone system, initiate tubular resorption of sodium and water, increasing the tubular concentration of oxalate.12 Tubular damage due to oxalate crystals attracts lymphocytes, causing interstitial nephritis, which ultimately leads to interstitial fibrosis and permanent loss of renal function.
When renal function decreases, serum oxalate levels rise.13 Research suggests that renal oxalate clearance decreases when GFR falls below 30-40 ml/min/1.73 m2 , causing elevated levels of oxalate and risk for further deterioration of renal function.4 Interestingly, a recent study showed an association between urine oxalate excretion and chronic kidney disease progression.14 Unfortunately, serum oxalate levels were not analysed. Systemic oxalosis, characterised by tissue damage in bones, skin, eyes, neurons, and heart due to deposition of oxalate crystals, may occur in patients with elevated levels of serum oxalate. Higher incidence rates of vascular events are seen in patients with systemic oxalosis, most likely explained by the toxicity of oxalate to the vascular endothelium.14 Oxalate nephropathy can manifest as both acute and chronic renal insufficiency. Though renal function may partially recover, more than half of all patients remain dependent on dialysis.15
DIAGNOSTICS
In a systematic review of patients with secondary oxalate nephropathy, a mean age of 56 years was identified, with men and women being equally affected.15
Urinalysis may reveal leukocyturia, haematuria, and mild proteinuria. Microscopic examination of urine shows birefringent calcium oxalate crystals in the urine in roughly one in four patients.15 Oxalate levels in both serum and urine may be elevated. Renal ultrasound may show nephrocalcinosis or nephrolithiasis, but as seen in our patients, this is not always the case. In renal biopsy, oxalate precipitations are seen in the tubular lumen, the interstitium, and the peritubulary capillaries (figure 1). Typically, the crystals are birefringent in polarized light.
TREATMENT
Treatment aims to reduce systemic oxalosis and to prevent oxalate nephropathy from developing. It relies primarily on decreasing enteric oxalate absorption by decreasing the amount of unbound oxalate in the enteric lumen. This can be achieved by reducing the amount of dietary oxalate and taking calcium supplements with meals. Foods with especially high oxalate content, such as rhubarb, spinach, purslane, Swiss chard, sorrel, and beetroot, should be avoided. Secondly, binding bile acids with cholestyramine reduces colonic permeability for oxalate, decreasing net absorption. Thirdly, supplementation of oxalate degrading bacteria such as Oxalobacter species, lowers intestinal oxalate levels and may decrease urinary oxalate excretion. Lastly, an ongoing trial investigates the potential of oral oxalate decarboxylase supplementation, which breaks down intestinal oxalate into carbon dioxide and formate.16
Fluid intake must be sufficient and dehydration should be avoided. High urine output lowers calcium oxalate supersaturation in the tubuli, decreasing the likelihood of renal crystal formation. Increasing pH of the urine by taking citrate, by correcting a metabolic acidosis, and by supplementing magnesium also reduces stone formation. Citrate also reduces calcium oxalate supersaturation by binding calcium, preventing oxalate crystal formation. Haemodialysis decreases serum oxalate levels. Patients who receive a renal transplant may also require haemodialysis in the initial days after surgery to prevent recurrence of oxalate nephropathy in the renal transplant. A living donor is preferred to minimize the chance of delayed graft function due to the injurious effects of the high oxalate burden in the transplanted kidney.1 Following kidney transplantation, hyperoxaluria may continue for years due to prolonged release of oxalate from the tissues.17 In this article, we explain that fat malabsorption due to exocrine pancreatic insufficiency can cause oxalate nephropathy. Since effective management is possible, early recognition is important. In clinical practice, patients with renal insufficiency who are at risk of developing pancreatic insufficiency due to malignancy or chronic pancreatitis (often secondary to alcohol abuse, smoking, or pancreatic duct obstruction), as well as patients who have had bariatric surgery, should be evaluated for oxalate nephropathy. Presumably, this diagnosis is often missed. Considering the increasing incidence of bariatric surgery, it is likely to occur more frequently in the near future. We hope to raise awareness for oxalate nephropathy secondary to fat malabsorption.
REFERENCES