KEYWORDS 
Acquired haemophilia A (AHA), malignancy
INTRODUCTION
Acquired haemophilia A (AHA) is a disease caused by spontaneous formation of auto-antibodies (inhibitors) against coagulation factor VIII. This can lead to severe and potentially life-threatening haemorrhages.
AHA is rare, with a reported incidence of 1.48 million/ year. Its frequency increases with age; the median age at diagnosis is 73.9 years.1,2 Aan underlying condition can be identified in about half of all cases. The most commonly associated conditions are autoimmune disorders, postpartum period, and solid or haematological tumours. In literature, the incidence rates for the coexistence of AHA and malignancy varies between 6-22%.3 Underlying malignancy is associated with worse prognosis of AHA.2 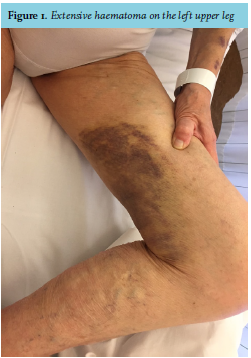 CASE REPORT
CASE REPORT
A 69-year-old woman presented at the emergency department with spontaneous bruising which started a week prior. Her medical history included metastatic breast cancer, for which palliative hormonal therapy (letrozole) was started, and non-small cell lung carcinoma, for which she had received chemoradiation therapy with curative intention. In addition, she had chronic obstructive pulmonary disease (COPD) and a mechanical aortic valve for which she used a vitamin K antagonist (acenocoumarol). Furthermore, she taking chlortalidone, metoprolol, losartan, Lyrica®, and oxycodone.
At physical examination, she had a blue, swollen right hand and a big haematoma on her left upper leg (figure 1). Laboratory testing showed a prolonged prothrombin time (PT) of 25.4 seconds and aPTT of 141 seconds (reference values: PT 9.5-12.5s, aPTT 20-30s). Platelet count was normal. After correction of the PT with prothrombin complex and vitamin K, the aPTT remained considerably prolonged (83s). Plasma mixing studies failed to correct the aPTT (57.2s after mixing, normal control 29.7s). The Rosner Index was 20.7, which indicates the presence of an inhibitor. The diagnosis of AHA was confirmed when there appeared to be an undetectably low concentration of factor VIII (< 0.01 IU/mL, normal value > 0.5 IU/mL) with a high titre of factor VIII antibodies (100.7 Bethesda Units, BU).
The day after admission, she started with high-dose prednisone (1 mg/kg) and was transferred to a haemophilia treatment centre. Given the high titre, it simultaneous administration of prednisone and cyclophosphamide was considered. However, the patient had recently been treated with chemoradiation therapy and therefore was already susceptible to serious infections. Starting cyclophosphamide, with great chance of additional toxicity, was considered too risky. Because of the extensive haematomas, the vitamin K antagonist was stopped. Given the risk of thrombosis of her aortic valve, we refrained from administration of a bypassing agent (like activated protrombincomplex or recombinant factor VIIa) and carefully followed the bleeding, which fortunately stabilized without the need for a bypassing agent. A few weeks after starting prednisone, the bleeding tendency was clinically reduced and the antibody titre dropped to 27 BU. The aPTT, however, remained prolonged (around 70s) and the factor VIII activity immeasurably low. Therefore, cyclophosphamide (100 mg a day) was started. However, because of intensive care admission with respiratory insufficiency due to a viral infection and an exacerbation of COPD, the immunosuppressive therapy was discontinued shortly after initiation. She received recombinant factor VIIa because of persistent bleeding after central line removal. After recovery from the infection, the cyclophosphamide was restarted at a dose of 50 mg a day. Three months after starting treatment, she reached remission with unmeasurable antibody titre and normalization of factor VIII. At this point, her vitamin K antagonist was restarted. She did not experience any thrombotic complications related to her mechanic aortic valve.
DISCUSSION
There is a clear association between acquired haemophilia A and malignancy. The presence of factor VIII antibodies is considered a paraneoplastic phenomenon. Due to the rarity of the disease, there is insufficient data to make statements about haemophilia A in specific types of cancer, but there seem to be differences in prevalence. Previous studies show that about a quarter of patients with AHA and malignancy had a lung tumour and one-fifth to a quarter had prostate cancer or a gastro-intestinal malignancy.1,4,5 In total, there are four known cases of patients with haemophilia A and breast cancer. There are no cases known of patients with a double tumour. If the diagnostic work-up of a new AHA patient shows evidence of malignancy, it is important to consider that tissue biopsy must be deferred until remission of AHA, as there is a high risk of bleeding complications.
Acquired haemophilia A should be considered in all patients with a recent onset of increased bleeding tendency and a prolonged aPTT with a normal PT and platelet count. Given the seriousness of the disease, additional tests must be done as soon as possible to confirm (or reject) the diagnosis. If the aPTT does not correct with a mixing test, this indicates the presence of a circulating inhibitor directed against one of the coagulation factors (VIII, IX, XI or XII), but the presence of lupus anticoagulans can also lead to a non-correcting mixing test. To make the diagnosis, the activity of the coagulation factors must be measured. If there is a coagulation factor VIII deficiency, the antibodies can be quantified by the Bethesda assay (the Nijmegen modification of the assay). Recent studies demonstrate there is a significant association between the height of the antibody titre and factor VIII activity on the time to achieve remission and on overall survival.6,7
Because of the rarity and severity of the condition, a haemophilia treatment centre must always be consulted and transfer should be discussed. Based on retrospective cohort studies and clinical research, guidelines and recommendations have been developed for the treatment of acquired haemophilia A. First advice is to strive for adequate haemostasis by means of recombinant factor VIIa (NovoSeven®) and activated prothrombin complex (Feiba®).8,9 No clinically relevant differences in efficacy and safety have been demonstrated and, if necessary, both products can be used simultaneously or sequentially if the effect of one product is insufficient.
As long as the antibody circulates in the blood, the risk of bleeding remains, and eradication of the inhibitor is therefore important. The development of antibodies against coagulation factor VIII is seen as an autoimmune phenomenon and is treated with immune suppressants. There are two treatment strategies: treatment with prednisolone only (1 mg/kg/day) or in combination with cyclophosphamide (1-2 mg/kg). Meta-analyses focusing on treatment results suggest that combination therapy is superior to prednisolone alone in achieving complete remission and disease-free survival. However, overall survival was not significantly better, probably because cyclophosphamide is associated with many severe and life-threatening side-effects like neutropenia-related infections.6,7,10 Therefore, the requirement to reduce the risk of severe bleeding always needs to be balanced with the risk of side effects, especially in the elderly and in patients with comorbidity that makes them more susceptible to infection. Treatment is monitored by determining aPTT, factor VIII activity and antibody titre. Meanwhile, attention should also be paid to the treatment of an underlying condition, if present.
In recent years, rituximab has been given more and more as second-line treatment. There are no randomized prospective studies comparing rituximab with other immunosuppressive treatments. Therefore, rituximab cannot yet be recommended as first line treatment.6,8,11
In conclusion, AHA is a rare disorder that should be considered as a haematological emergency. It is idiopathic in half of all patients, and in the other half, there is a clear association with an underlying condition, for instance a malignancy. Since few data are available on treatment and prognosis, registration of patients is of great importance to gain more insight into this disorder, and its etiology, treatment, and prognosis.
REFERENCES