
KEYWORDS
Adrenocortical carcinoma, hypercortisolism, metabolic syndrome
CASE REPORT
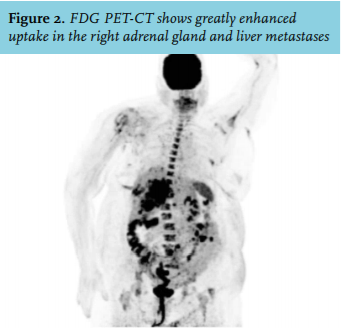
A 75-year-old woman was referred to our hospital because of malignancy with liver metastases suspicious for hepatocellular carcinoma, as determined by CT scan and biopsy. She was known with type 2 diabetes since 2008, until recently well-regulated with dietary measures only. However, in the past months, she was admitted to the referring hospital twice because of congestive heart failure and hyperglycaemic deterioration with an Hb1ac level of 116 mmol/mol, for which metformin and insulin glargine were started. She was taking several antihypertensive drugs (metoprolol, spironolactone, amlodipine and valsartan). Her symptoms at presentation included severe muscle weakness, leading to wheelchair dependency in one month. On clinical examination the patient appeared frail with proximal muscle weakness and haematomas localised at her right breast and arm after a recent collapse. Centripetal obesity and peripheral oedema were present. Blood pressure was 170/95 mmHg, pulse rate 86 bpm and her BMI was 34.3 kg/m2. Laboratory testing showed: potassium 1.6 mmol/l (reference values 3.5-5.1), sodium 148 mmol/l (136-145), fasting glucose 14.6 mmol/l (4.0-6.1), phosphate 0.19 mmol/l (0.80-1.40), AST 109 U/l (< 31), LD 1381 U/l (< 247) and gamma-GT 964 U/l (< 38). A severe metabolic alkalosis was seen with a pH of 7.61 (7.35-7.45) and a bicarbonate of 47 mmol/l (21-27). Re-evaluation of the CT scan performed elsewhere identified a right-sided adrenal mass invading the right renal vein, inferior vena cava and right liver lobes (figure 1), suspicious for adrenocortical carcinoma (ACC) with liver metastases. Pulmonary embolisms were also detected. Additional hormonal testing yielded a serum cortisol level of 2180 nmol/ (200-800), a midnight salivary cortisol level of 339 nmol/l (< 2.8), elevated urinary cortisol excretion at 6232 nmol/24 hours (< 133) and a suppressed adrenocorticotrophic hormone (ACTH) concentration of 1.2 pmol/l (1-55). The serum steroid profile showed increased steroid precursor levels, including androstenedione, progesterone, 17-hydroxyprogesterone, testosterone and oestradiol, while aldosterone levels were suppressed; 24-hour urinary excretion of (nor)metanephrines was not elevated. High uptake in the adrenal tumour and liver metastases was shown on the 18F-fluoro-deoxyglucose positron emission tomography (FDG PET)-CT (figure 2). After revision by our pathologist, the liver biopsy was concordant with an ACC with positive Melan-A and synaptophysin staining and a Ki67 index of 20%. In summary, our patient was diagnosed with a stage IV ACC producing cortisol, androgen and oestrogen. Because of her poor performance status she was deemed ineligible for surgery, mitotane or intensive chemotherapeutic treatment. Cortisol excess was treated with metyrapone combined with hydrocortisone suppletion (block and replace therapy). Spironolactone was increased from 50 mg once daily to 150 mg twice daily combined with potassium supplements to treat hypokalaemia. Three days after starting block and replace therapy a biochemical response was seen as evidenced by normalisation of the blood pressure, a lower insulin dose requirement and resolution of alkalosis. Treatment with low-molecular-weight heparin was started for pulmonary embolism. The patient was discharged to a palliative care facility.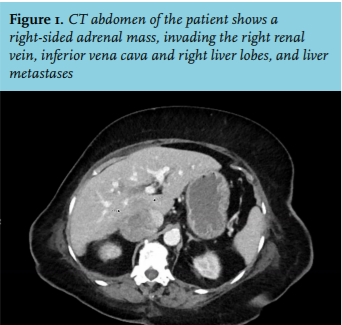
DISCUSSION
ACCs are rarely seen with an incidence of 1-2 per million persons/year. Less than 5% of adrenal incidentalomas appear to be ACCs.1 In adults, ACCs typically present in the fourth to fifth decade as a sporadic tumour, more often in women than in men (male female ratio 2:1). Approximately 60% of ACCs are functional causing a clinical syndrome as a result of an excess of cortisol, androgens, oestrogens or rarely aldosterone. ACTH-independent Cushing’s syndrome caused by autonomous cortisol production is most frequently seen (45%) whereas less than 10% of patients present with virilisation alone.2 The morbidity of Cushing’s syndrome can be classified into metabolic, cardiovascular, musculoskeletal, infectious, reproductive, dermatological and neuropsychiatric manifestations.3 Glucose intolerance is primarily due to stimulation of gluconeogenesis by cortisol, direct suppression of insulin release and peripheral insulin resistance caused by progressive centripetal obesity.4 Direct stimulation of the mineralocorticoid receptor by glucocorticoid excess, exceeding the capacity of 11-beta-hydroxysteroid dehydrogenase type II in the kidney, causes hypertension, hypokalaemia and oedema.5 This pseudoaldosteronism was also present in our patient, also evidenced by the metabolic alkalosis. Thromboembolic events are frequently seen, caused by both increased plasma concentrations of clotting factors with activation of the coagulation cascade and inhibition of fibrinolysis.6 Due to clustering of cardiovascular risk factors, patients are at risk for cardiovascular events.7 Proximal muscle weakness and osteoporosis are caused by the catabolic effects of hypercortisolism on skeletal muscles and bone, respectively. An increased risk of infections is the result of immunosuppressive effects. An important clinical clue to the presence of hypercortisolism is the simultaneous development and increasing severity of several of the aforementioned symptoms causing considerable morbidity.
Non-functional ACCs are often discovered as incidentalomas. When an ACC is suspected, hormonal evaluation should always be performed, including fasting blood glucose, serum potassium, cortisol, ACTH, 24-hour urinary free cortisol, dexamethasone suppression test, aldosterone, renin, dehydroepiandrosterone-sulphate (DHEAS), androstenedione, testosterone, 17-hydroxyprogesterone and, on indication, oestradiol in men and postmenopausal women. It is also recommended to measure plasma or urinary (nor)metanephrines to exclude pheochromocytoma.1 A CT scan can usually distinguish adenomas from ACCs; however, with magnetic resonance imaging local invasion of veins can be identified more precisely. Most ACCs are greater than 4 cm when discovered. Since the liver, lungs, lymph nodes and bones are the most common sites of distant metastases, a CT thorax/abdomen is included in the diagnostic work-up. The role of FDG PET-CT needs to be further determined but studies using PET/CT to compare malignant lesions with adenomas show a high sensitivity, specificity, positive predictive value, and negative predictive value of 100%, 98%, 97%, 100%, respectively.8 Fine-needle aspiration biopsy is only recommended when an adrenal metastasis of another malignancy is suspected. For histopathological examination, the Weiss system is most commonly used.9 Ki67 proliferation index as determined using immuno/ histochemistry has consistently been related to prognosis.10 Staging of ACC patients is performed with the ENSAT 2008 staging system.1
At presentation, approximately 50% of adult patients have advanced stage III or IV disease.11 In a palliative or metastasised setting mitotane monotherapy can be considered in patients with low-grade disease, late recurrence or those unfit to undergo chemotherapy. However, a survival benefit has not been proven.12 Patients undergoing mitotane treatment can experience substantial toxicity, mostly gastrointestinal and neurological, also due to the small therapeutic range of this drug. For patients with aggressive tumours and WHO performance score 0-1, combination chemotherapy including etoposide, doxorubicin and cisplatin with mitotane is advised based on the FIRM-ACT trial that revealed a slight progressionfree survival benefit of this regimen over streptozocin with mitotane.13 In case of Cushing’s syndrome, tumour debulking or treatment with metyrapone or ketoconazole, next to the cortisol-lowering effects of mitotane, can be necessary to block adrenocortical cortisol synthesis.2 Suppletion with hydrocortisone might be necessary on indication. Radiation therapy can be helpful in palliating pain. Overall, the prognosis of ACC is poor with a five-year survival of approximately 55 to 65% for early stage and 10 to 25% for advanced stage disease.14
Our case shows a rare cause of worsening of a pre-existing metabolic syndrome with sudden dysregulation of hypertension and diabetes as the clue to the underlying cortisol-producing ACC. Cushing’s syndrome is rare and the diagnosis is often delayed because part of its morbidity, namely the metabolic syndrome, has a high prevalence in the general population. The typical combination of Cushing’s syndrome features or sudden worsening of symptoms should prompt the clinician to consider hypercortisolism as underlying cause. Since severe hypercortisolism can be lethal, it is essential to start appropriate treatment quickly.
DISCLOSURES
All authors declare no conflict of interest. No funding or financial support was received.
REFERENCES