KEYWORDS
TPP, hypokalaemia, periodic paralysis
INTRODUCTION
Periodic paralysis is an intriguing medical emergency, with otherwise healthy young individuals presenting with acute painless muscle weakness due to potassium shifts. Two subtypes of hypokalaemic periodic paralysis can be distinguished: a hereditary and an acquired form.1 In Western countries, autosomal dominant hypokalaemic periodic paralysis (HPP) is common, with a prevalence of 1 in 100,000.2 This is caused by mutations of voltagedependent calcium, sodium or potassium channels of the skeletal muscle membranes, with symptoms typically beginning in the first or second decade of life. Although clinical features of sporadic hypokalaemic paralysis are similar to HPP, de novo mutations were identified in a minority of patients.3
In Asia, the acquired form is common although, to our knowledge, its exact prevalence has not been reported. Due to its association with hyperthyroidism, this form is called thyrotoxic periodic paralysis (TPP), and it usually occurs after the second decade of life. TPP has an incidence of 1.8% among patients with hyperthyroidism in Asia, whereas rates are estimated to be around 0.1% in the West and usually include patients of Asian background.4-6 Only few cases of Caucasians with TPP have been described in the literature.7,8 In contrast to primary autoimmune hyperthyroidism occurring predominantly in females, TPP mostly affects males (male-to-female ratio 26:1).9,10 The average age of onset is around 30 years which, unsurprisingly, coincides with the average age of onset of Graves’ disease.9-15
Because this condition is rare, it may be frequently overlooked and misdiagnosed, while recognition and therapy could prevent deleterious outcomes. Mortality rates are unknown, but TPP may explain a portion of sudden cardiac deaths among young Asians.16 With increasing migration it is likely to be seen more frequently in Western countries. Here, we briefly report three cases of TPP patients in the Netherlands, and review the literature.
HISTORY
The combination of muscle weakness and hyperthyroidism was first described in 1902.17 In 1931, four cases of hyperthyroidism and attacks of muscle weakness were described in the English literature.18 Treatment of the hyperthyroidism resulted in resolution of the periodic paralysis, but this recurred in one case during a relapse of hyperthyroidism.18 Subsequently, the decrease in serum potassium levels was found to be proportional to the severity of the paralysis.4
GENETICS AND PATHOPHYSIOLOGY
Genetic predisposition only combined with hyperthyroidism is thought to induce TPP (figures 1 and 2).19 Observations indicating a genetic predisposition include a higher prevalence among Asians, and the association of paralysis with recurrence of hyperthyroidism. Because of the clinical similarity between TPP and HPP, candidate genes that cause HPP were initially evaluated. These included genes that encode for channels needed for membrane potential stability or excitability, including the voltage-gated skeletal sodium channel Nav 1.4 (SCN4A), calcium channel Cav 1.1 (CACNA1S) and less frequently the inward rectifier potassium channel Kir2.1 (KCNJ2).20-24 Despite clinical resemblance, no mutations of these genes have been identified in patients with TPP.25,26 However, single-nucleotide polymorphisms (SNP) in the CACNA1S region (encoding for Cav 1.1) have been discovered. Expression of this calcium channel may alter thyroid hormone action.26
Ryan and colleagues identified a mutation of KCNJ18, which encodes for the potassium channel Kir2.6.27 Kir2.6 associates with other Kir2.x subunits, such as Kir2.1, forming a homotetrameric complex and, thereby, alters inward potassium trafficking.28 Potassium fluxes are required for stabilising the resting membrane potential. Interestingly, Kir2.6 contains a thyroid responsive element that may regulate gene transcription (figure 1). Decreased inward currents of potassium predispose the sarcolemma to paradoxical depolarisation, which in turn may lead to muscle paralysis.27,29 Very recently one c-terminus mutation of Kir2.6 (D252N) was shown to decrease potassium currents by 34%.30 However, the KCNJ18 mutation is only present in 25-33% of TPP patients from France, United States, Brazil and Singapore, and it was absent in Chinese and Thai subjects.27 Therefore, the majority of TPP patients have as yet unidentified disease-causing mutations. Genome-wide association studies identified the genetic variant rs312729, which is located downstream and may affect Kir2.1 expression, as a new susceptible locus for TPP in Thai patients.31 This association, but also other variants such as rs312691, were further confirmed in Chinese and Korean TPP patients.32-34 In Chinese patients, Song and colleagues identified the genetic variant rs312736, which mapped to CTD-2379E21.1 that encodes for a noncoding RNA and regulates KCNJ2 expression, to be associated with TPP.35 With these recent findings, it seems the inward rectifier potassium channels play a pivotal role in the predisposition of TPP.
These findings, however, do not explain why TPP patients have attacks of hypokalaemia during hyperthyroidism. Further evidence indicates that increased activity of the Na+/K+-ATPase may be the link (figure 1). Indeed, Na+/ K+-ATPase abundance and activity is exaggerated in TPP as compared with healthy control subjects, but also as compared with non-TTP patients with primary hyperthyroidism.36 This is attributed to a direct effect of hyperthyroidism, but also through an increased expression of β2-adrenergic receptors.37 In addition, insulin, adrenergic stimulation, androgens or exercise further increase Na+/K+-ATPase activity.38-40 This explains why these factors may predispose to an attack in TTP patients.9,10,12,41 Impaired potassium efflux caused by mutations of the inward rectifier potassium channels, combined with increased Na+/K+-ATPase activity may lead to paradoxical depolarisation with muscle inexcitability and eventually paralysis (figure 1).42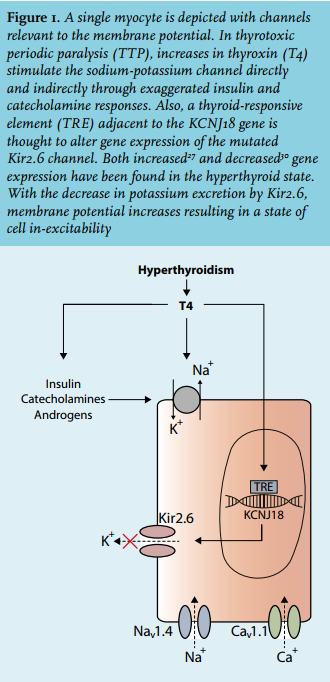
CLINICAL FEATURES
Hypokalaemic paralytic attacks only occur during hyperthyroidism (figure 2). Prodromes include muscle aches, stiffness, weakness or cramps 1 hour to 3 days before paralysis.4,12 Before presentation, most patients have experienced less severe muscle weakness that resolved spontaneously. Attacks usually begin with weakness of the proximal muscles of the lower extremities, and may progress to tetraplegia, with the degree of muscle weakness corresponding to serum potassium levels.15 No correlations with serum T3 or T4 levels have been found. Bowel and bladder function, facial expression, swallowing and respiration are usually unaffected.12,15 A precipitating factor for an attack could be identified in 34% of patients.9 Precipitating factors include high carbohydrate ingestion, alcohol, infection (mainly respiratory or urinary tract infections have been described), excessive exercise and use of β2-adrenergic bronchodilators (figure 2).9,10,12,41 Furthermore, an attack was inducible by glucose loading in a minority of patients.9 Patients usually experience attacks in the early mornings, and during warm seasons.4,10 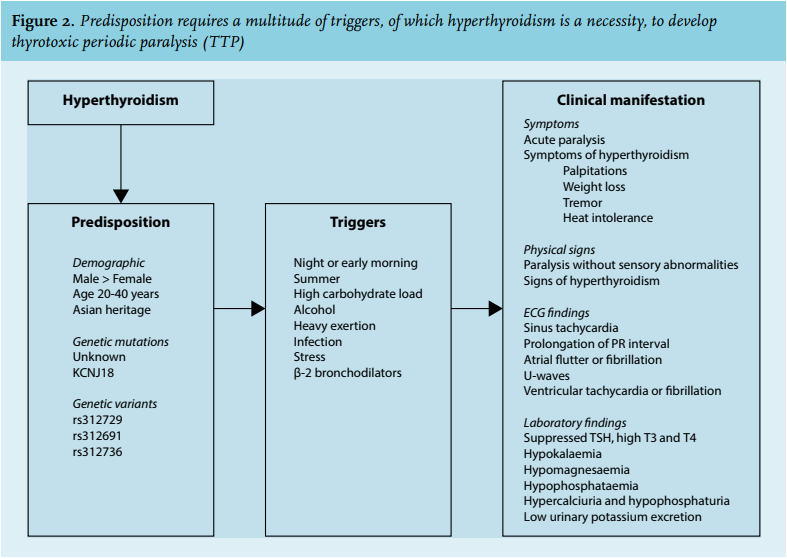
Laboratory findings
At presentation, laboratory findings include very low levels of serum potassium (on average 2.0 mmol/l), without acid-base disturbances.9,10,12-15 Renal excretion of potassium is also low, excluding renal potassium wasting. Some reports also mention low serum phosphate and magnesium levels, which also tend to resolve spontaneously.12-14 A high urinary calcium-to-phosphate ratio has been proposed to distinguish TPP patients from those with HPP.13
Furthermore, suppressed serum thyroid-stimulating hormone (TSH) with high T4 and T3 levels are pathognomonic for TPP, and distinguish TPP from HPP patients. Free T4 levels are similar to those in non-TTP patients with primary hyperthyroidism.10,12-14 Graves’ disease is diagnosed in the majority of TPP patients, which is also unsurprisingly the main cause of hyperthyroidism in general. However, other causes such as toxic adenoma, toxic multinodular goitre, thyroiditis, and TSH-producing pituitary tumours have been reported in patients with TPP.9,12,43-45 Notably, 17-85% of TPP patients have mild signs of hyperthyroidism at the time of diagnosis,9,10,12 and TPP is the first presentation of hyperthyroidism in about 80% of the cases.46
Electrocardiogram
In TPP, both hyperthyroidism and hypokalaemia may cause cardiac arrhythmias that will be apparent on the electrocardiogram (ECG). Sinus tachycardia, and also atrial fibrillation or flutter, are common findings in hyperthyroidism.47 During an attack, hypokalaemia slows repolarisation, prolongs the refractory period and thereby predisposes to arrhythmias. This is exacerbated as ventricular repolarisation is also lengthened in hyperthyroidism;48 the ECG may show supraventricular or ventricular ectopic beats, prolonged PR interval, and U waves.14 Because the risk of serious ventricular arrhythmias is high, including life-threatening ventricular tachycardia or fibrillation,16 cardiac monitoring is necessary until hypokalaemia is corrected.
TREATMENT
To prevent major complications, such as cardiac arrhythmias, the initial therapy of TTP should be directed to the correction of the hypokalaemia. Some report a shortened recovery time of the paralysis by potassium supplementation,49 while others do not.12 Because total body potassium levels are normal, aggressive treatment may result in rebound hyperkalaemia, which occurs in approximately 40-60% of TPP.12,14 Therefore, low-dose potassium supplementation (10-20 mmol/h) is recommended, unless there are serious cardiovascular complications.49,50 Propranolol (a nonselective beta-blocker) has been reported to normalise serum potassium within two hours without rebound hyperkalaemia. The mechanism of action of this drug is the reversal of the adrenergic stimulation of the Na+ /K+-ATPase and reduction of the conversion of T4 to T3 in the liver resulting in lower circulating T3 levels.51,52 Propranolol may be continued until a euthyroid state is achieved. However, awaiting correct diagnosis, potassium supplementation should also be initiated, with frequent monitoring of serum potassium levels. Meanwhile, patients should be advised to avoid precipitating factors, such as alcohol, high carbohydrate ingestion and excessive exercise.
Initially, thyrotoxicosis should be treated with antithyroid drugs. Block and replace therapy is recommended because of better control of thyroid hormone levels compared with a titration strategy.53 Next, definitive treatment of the hyperthyroidism is required to prevent relapse of paralytic attacks, which may occur in 62% of patients in the first three months after diagnosis.10 Antithyroid drug treatment was associated with relapse of TPP, mostly during withdrawal or tapering of medication.46 Radioactive iodine or surgical treatment is the most definitive treatment, thereby avoiding the risk of recurrence of TPP. When thyroid hormone replacement is initiated because of iatrogenic hypothyroidism, utmost care in dosing is essential, as even slight overtreatment can lead to TPP relapse.4
ILLUSTRATIVE CASE REPORTS
Table 1 shows the characteristics and main clinical findings of three patients with TPP who presented to the medical emergency department. All three patients were male, of Asian descent and presented with complaints of muscle weakness. One patient had experienced muscle weakness previously but this had resolved spontaneously. Possible triggers were high carbohydrate and alcohol consumption. Physical examination revealed paresis of the extremities in all three cases, while sensation was intact. In two cases, severe tetraparesis was present. Serum potassium levels were low to extremely low (1.4 to 2.4 mEq/l), without metabolic abnormalities. With low urinary potassium excretion, renal causes of hypokalaemia were excluded. All cases had abnormal ECG findings, corresponding with hypokalaemia. Interestingly, there were no prodromes indicating hyperthyroidism in all three cases. Serum TSH was only measured after correction of serum potassium, as causes of low potassium and paralysis were evaluated. Therefore, none were treated with propranolol at admission, and two patients had rebound hyperkalaemia after potassium supplementation. Graves’ disease was diagnosed in all three cases based on high anti-TSH receptor antibody levels and diffuse thyroid uptake of radioactive iodine in combination with suppressed serum TSH and elevated serum free T4 levels. In all three cases the paralysis disappeared within hours of treatment, and patients were subsequently also treated for their primary hyperthyroidism. One patient (case 3) required thyroidectomy because of a recurrence of the paralysis shortly after initiation of treatment, which was likely triggered by alcohol consumption. Definitive treatment was advised to the other two patients, but case 1 declined and case 2 was lost to follow-up. 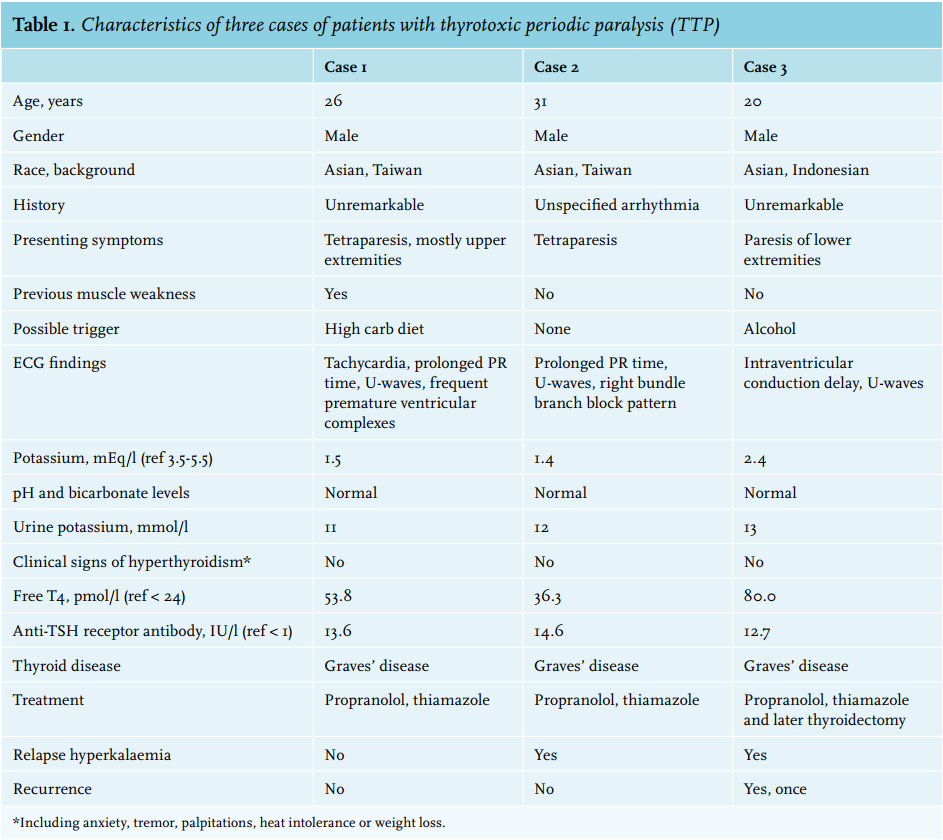
CONCLUSION
Sudden onset of paralysis with hypokalaemia, especially in Asian males, should trigger clinicians to consider acquired periodic paralysis. In many cases, TPP may be the first presentation of hyperthyroidism. If left untreated, recurrence of hypokalaemia is likely, and this may cause life-threatening arrhythmias. Treatment relies on supplementation of potassium chloride, nonselective beta-blockers and ultimately treatment of the underlying hyperthyroidism. Because total body levels of potassium are normal, frequent measurements of serum potassium levels and adjustment of the supplementation rate are needed to avoid rebound hyperkalaemia. With globalisation, clinicians in the Western world are more likely to encounter patients with TPP.
DISCLOSURES
None of the authors received financial support in relation to the studies as described in this manuscript.
REFERENCES