
KEYWORDS
Congenital adrenal hyperplasia, adrenal incidentaloma, urinary steroid profile
INTRODUCTION
An adrenal incidentaloma (AI) is an asymptomatic adrenal mass detected on imaging not performed for suspected adrenal disease.1 In radiological series the prevalence is estimated at 2-4% in middle-aged patients, increasing to 10% in 70-year-olds.2 Guidelines recommend repeated imaging studies to assess the risk of malignancy and biochemical evaluation to identify possible hormonal activity.1,3 Adrenalectomy is indicated in case of hormone excess or radiological characteristics suggestive of malignancy.1
We present a patient with an AI who initially seemed to meet the criteria for adrenalectomy. Additional analysis, however, revealed an underlying aetiology for which medical treatment was successfully instituted.
CASE REPORT
A 43-year-old male suffered from progressive fatigue and myalgia for more than a year. Evaluation by his general practitioner did not result in a diagnosis. The patient decided to visit a commercial clinic for an MRI scan which revealed a tumour of 5.2 x 4.4 cm in the left adrenal gland. Subsequent analysis elsewhere excluded the presence of hormonal hypersecretion. An unenhanced CT scan was performed, which demonstrated a homogenous lesion in the left adrenal gland with a radiodensity of 19 Hounsfield units and a normal appearing contralateral adrenal gland (figure 1). A ‘wait-and-scan’ strategy was proposed. The patient, however, opted for surgical removal and was referred to our hospital.
His past medical history was uneventful. Besides fatigue and myalgia he reported frequent headaches and night sweats without fever, but had no other complaints. He and his wife were involuntarily childless. He recalled being taller than most of his peers at the age of ten, but ending up as one of the shortest by the end of puberty.
No abnormalities were found at physical examination. He was 1.70 m in height and weighed 69 kg. Laboratory analysis demonstrated a normal complete blood count, electrolytes, glucose, renal and liver function tests and plasma metanephrines. The results of additional hormone measurements showed: cortisol 350 nmol/l (at 14.00 h), ACTH 27 ng/l (reference range: < 23 ng/l), 17OH-progesterone (17-OHP) 426 nmol/l (4.0-12.0 nmol/l), androstenedione 14 nmol/l (2.6-7.2 nmol/l), DHEAS 3.1 µmol/l (2-7 µmol/l), testosterone 13 nmol/l (16-40 nmol/l), luteinising hormone 3.61 U/l (2.1-11.2 U/l), follicle-stimulating hormone 6.68 U/l (1.8-7.2 U/l), plasma renin activity (PRA) 1.6 nmol/l/h (0.10-2.35 nmol/l/h). Urinary steroid profiling revealed a markedly increased excretion of pregnanediolone, pregnanetriol and pregnanetriolone. Intravenous administration of 250 µg tetracosactide resulted in a peak serum cortisol and 17-OHP of 415 nmol/l and 997 nmol/l, respectively.
We diagnosed congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency. DNA analysis demonstrated compound heterozygous mutations of the CYP21A2 gene (c.518T>A (p.Ile173Asn) and c.710T>A, c.713T>A, c.719T>A (p.lIe237Asn), (p.Val238Glu), (p.Met240Lys)), which are associated with the simple virilising form of classic CAH.4 Ultrasound examination excluded the presence of testicular adrenal rest tumours.
Dexamethasone 0.5 mg once daily was started, resulting in prompt resolution of all his symptoms and biochemical normalisation of the adrenal steroid precursors. The tumour size decreased (4.4 x 2.9 cm) after one year of treatment (figure 1). Dexamethasone was switched to hydrocortisone 10 mg thrice daily with maintenance of excellent clinical and biochemical control.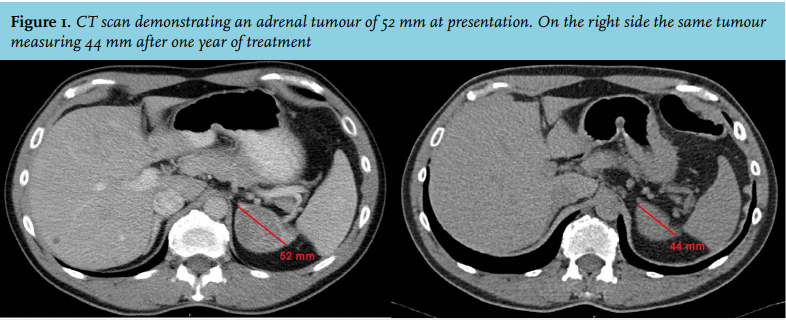
DISCUSSION
We present a patient with a large adrenal tumour caused by newly diagnosed classic CAH due to 21-hydroxylase deficiency (21-OHD). Treatment with glucocorticoids was successful and adrenalectomy was thus avoided.
CAH represents a group of autosomal recessive inherited disorders in steroid biosynthesis. Approximately 95% of cases are due to 21-OHD.5 The most severe form is classic CAH, characterised by adrenal insufficiency with or without aldosterone deficiency (i.e. salt-wasting or simple virilising phenotype, respectively), disorders of sexual development with genital ambiguity in girls, short stature and infertility. Mild 21-OHD results in non-classic CAH, in which genital ambiguity and cortisol deficiency are absent. Patients with non-classic CAH often have manifestations of hyperandrogenism, such as early pubarche, acne, hirsutism or oligomenorrhoea/amenorrhoea. The prevalence of CAH and non-classic CAH is estimated at 1 in 10,000-15,000 and 1 in 500-1000 live births, respectively.5 Neonatal screening for 21-OHD has been performed in the Netherlands since 2000. Consequently, the risk of missing a diagnosis of classic CAH during childhood is extremely low nowadays.6
Adrenal tumours have been reported in 11-82% of patients with CAH, the development of which is probably enhanced by prolonged ACTH stimulation of the adrenal cortex.7,8 About 45% of these tumours are unilateral, which means that other, as yet unknown, factors contribute to adrenal tumour development in patients with CAH.8 It has been estimated that among patients with an AI only 0.8% are caused by CAH. This raises the question as to when an underlying diagnosis of CAH should be ruled out. In general, guidelines on AI management recommend adrenalectomy when the diameter of the tumour exceeds 4-6 cm, as the risk of adrenocortical carcinoma increases with size.1 Direct application of this recommendation to our patient would have resulted in an unnecessary adrenalectomy, including the risk of provoking a perioperative Addisonian crisis. Therefore, before adrenalectomy is considered in a patient with AI, the medical history should be checked for symptoms and signs suggestive of CAH such as premature pubarche, short stature and hyperandrogenism.8 In addition, sex steroids and their precursors (including 17-OHP) should be measured, as recommended by the recent European Society of Endocrinology guideline on AI management as well as the European Society for Medical Oncology guideline on adrenal cancer.3,9 High serum levels of 17-OHP are suggestive of CAH, which could be further evaluated by an intravenous tetracosactide stimulation test and, finally, confirmed by genetic analysis. Notably, moderately elevated levels of 17-OHP might also accompany a large adrenal adenoma or an adrenocortical carcinoma.10 Thus, the specificity of an elevated serum 17-OHP for CAH is limited in the presence of AI.8 Urinary steroid profiling (USP) by gas chromatographymass spectrometry might overcome this problem by demonstrating increased levels of pregnanetriol, pregnanediolone and pregnanetriolone with low levels of cortisol metabolites in a patient with CAH.11 Another advantage of USP is that it provides a comprehensive evaluation of the steroid biosynthesis which enables detection of not only 21-OHD, but also all other causes of CAH. In addition, recent evidence suggests that USP can distinguish between adrenal adenoma and adrenocortical carcinoma.12,13 Thus, USP serves several diagnostic purposes and seems to be a promising first-line test in AI analysis.
In conclusion, adrenal tumours are common in CAH, but CAH as the cause of AI is rare. CAH should be excluded in case of clinical suspicion or when adrenalectomy is considered. Urinary steroid profiling is a useful diagnostic tool in patients presenting with AI.
DISCLOSURES
No conflict of interest declared by the authors.
REFERENCES