
KEYWORDS
Autosomal dominant hypocalcemia, calcium-sensing receptor, hypocalcemia
INTRODUCTION
Chronic hypocalcaemia is a frequent problem and can be life-threatening. Most patients report paraesthesia, but hypocalcaemia can also cause muscle cramps, seizures and cardiac arrhythmias. Long-term complications of chronic hypocalcaemia include tissue calcifications in the brain and kidneys, cataract and osteoporosis. Parathyroid hormone (PTH) is responsible for minute-tominute regulation of the plasma calcium.1,2 Hence, causes of hypocalcaemia are classified according to PTH concentration (table 1).
The parathyroid cells express calcium-sensing receptor (CaSR), a G-protein-coupled receptor providing a negative feedback system to the calcium-PTH axis.3-5 CaSR is mainly located in parathyroid glands and the renal tubule. By sensing ionised calcium, the CaSR regulates PTH release and PTH-independent calciuresis. Gain-of-function mutations in the CaSR gene shift the calcium-PTH axis to a lower set point. The mutant CaSR becomes activated by a low ionised serum calcium concentration and subsequently inhibits PTH release. Autosomal dominant hypocalcaemia (ADH) is a syndrome characterised by an inappropriately low PTH according to a symptomatic hypocalcaemia and a relative hypercalciuria.1,4,5 A small subgroup of ADH patients develop additional renal loss of sodium, chloride and magnesium, resulting in hyperreninaemia, hyperaldosteronism, hypokalaemia and metabolic alkalosis. Most of them present during childhood, which is diagnosed as Bartter’s syndrome type 5.4,6
We present a family of a father and his sons diagnosed with ADH caused by a new mutation in the CaSR gene.
CASE REPORT
A 47-year-old bald man (index, patient I1 ) was seen with complaints of paraesthesia in both limbs. No Chvostek sign could be provoked. His past medical history included autoimmune hypothyroidism. Blood analysis showed a serum calcium of 1.95 mmol/l at presentation. His phosphate was 0.81 mmol/l and PTH was low 6.6 ng/l (table 2). The calciuresis was high at 7.6 mmol/24 h. Patient I1 was initially diagnosed with idiopathic hypoparathyroidism and treated with calcitriol. We performed genetic testing by polymerase chain reaction (PCR) and Sanger sequencing of the coding regions and intron boundaries of the CaSR gene. Genetic analysis by PCR amplification and DNA sequencing revealed a new mutation in the gene coding for CaSR: c.2195A>G, p.(Asn732Ser) in exon 7. Since patient I1 is a heterozygous carrier, genetic counselling was recommended with his two sons.
One son (patient II1 ) was analysed at the age of 25. He reported a tingling sensation in both hands. He smoked 25 cigarettes a day. Physical examination revealed no Chvostek sign, hypertension 140/70 mmHg, and a remarkable baldness. PCR of the CaSR gene exposed the same mutation as patient I1 had. Lab results of patient II1 showed hypocalcaemia 1.92 mmol/l with a normal concentration of albumin, phosphate and PTH. Calciuresis was also high-normal at 6.3 mmol/24 h (table 2).
The youngest son (patient II2 ) is asymptomatic and still has all his scalp hair. No mutation in the calcium-sensing receptor could be found.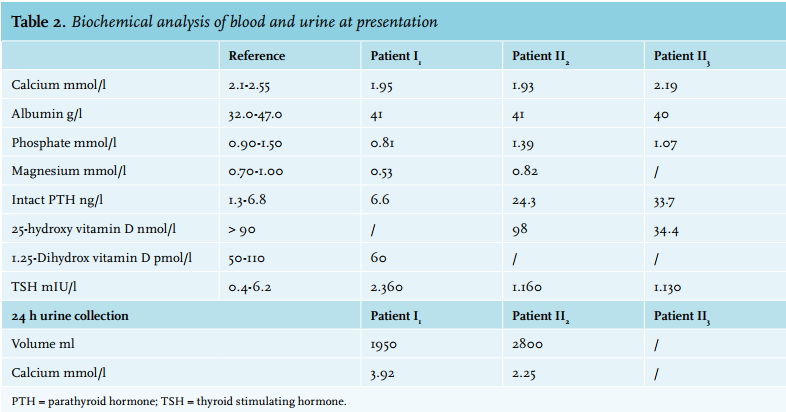
DISCUSSION
Activating mutations of the CaSR gene can cause symptomatic hypocalcaemia. ADH has a wide clinical spectrum.5,7 As in our cases, most patients report paraesthesia in the limbs, but chronic severe hypocalcaemia might be life-threatening.1,4,5,7 The phenotype is determined by the calcium level. Signe et al. found that the severity of clinical neurological symptoms is inversely related to serum calcium levels.7
The CaSR gene is located on chromosome 3q13.5,8. The first activating mutation was described by Pollak et al. in 1994.9 The CaSR mutation database (http://www. casrdb.mcgill.ca) contains more than 40 known activating mutations in CaSR gene.5 Also several recent case reports have demonstrated new activating mutations.8,10 Genetic analysis in patient I1 and patient II1 revealed another new activating mutation: c.2195A>G, p.(Asn732Ser) in exon 7. Because this mutation has not been described before, it is classified as ‘variant of uncertain significance’. In silico analysis of the mutation shows that substitution of asparagine by serine is most probably pathogenic. Most ADH patients are heterozygous. One family is known with a homozygote mutation, but this is not associated with a more severe phenotype.5
Remarkably, our affected patients are completely bald. Alopecia is a known symptom occurring in polyglandular autoimmune syndrome type I (PGA I).1,5 Although patient I1 has an autoimmune hypothyroidism, no Addison’s disease, mucocutaneous candidiasis, keratopathy, vitiligo, parietal cell atrophy, insulindependent diabetes mellitus or autoimmune hepatitis is known in this family. Therefore, it is unlikely that the alopecia in our patients fits into an autoimmune syndrome. Moreover, PGA I is inherited as an autosomal recessive trait and has a childhood onset.1,5 Another hereditary syndrome is an autosomal recessive mutation in the hairless gene located on chromosome 8p12.11
However, neither a biopsy, nor genetic counselling towards the hairless gene was performed. Most probably, a mutant gene coding for alopecia is located close to the CaSR gene and was simultaneously inherited.
Treatment of hypocalcaemia is symptomatic. Hypocalcaemia is treated by calcium supplementation. Calcitriol or alphacalcidol is added to stimulate intestinal calcium absorption. Normalisation of hypocalcaemia can cause a rise in hypercalciuria resulting in kidney stone formation. Therefore, treatment with active vitamin D and calcium supplementation should be performed carefully to prevent symptoms and arrhythmias, but also to prevent possible long-term complications of this treatment. Each patient requires regular monitoring of serum calcium, calciuria and renal function. Adding hydrochlorothiazide can limit calciuria.4
Ideally, the activated CaSR should been blocked to provide a more pathophysiological therapy. Calcilytic drugs are being studied to block the CaSR and thus provide a more pathophysiological approach in treating these patients. This could be a very promising novel therapeutic approach for ADH.12
In conclusion, we present a father and his son who were diagnosed with ADH. Their activating mutation in the CaSR gene leads to symptomatic hypocalcaemia. In both family members a new mutation in the CaSR was found. Possibly, a polymorphism of a gene related to alopecia is located near the CaSR gene and is also dominantly inherited.
DISCLOSURES
The authors declare no conflicts of interest.
REFERENCES