KEYWORDS
Diabetes mellitus, HNF1A, MODY
INTRODUCTION
Diabetes mellitus is a worldwide disease associated with microvascular and macrovascular complications and still has an increasing prevalence and incidence.1
Although most patients with diabetes mellitus have either type 1 diabetes (~10%) characterised by primary insulin deficiency due to autoimmune β-cell destruction, or type 2 diabetes (~85%) characterised by insulin resistance and relative insulin deficiency, other types of diabetes mellitus do exist. In contrast to the more complex multifactorial origin of type 1 and type 2 diabetes mellitus, some of the less prevalent forms of diabetes have a monogenetic origin. Of these, maternally inherited diabetes and deafness with a prevalence of around 1% in de diabetic population,2 and maturity onset diabetes of the young (MODY) constitute the most important diabetes subtypes.
MODY comprises a distinct group of monogenic and autosomal dominant inherited forms of diabetes mellitus due to β-cell dysfunction with onset at a young age. It may be difficult to distinguish from late onset type 1 diabetes and early onset type 2 diabetes, due to the absence of clear distinguishing features at diagnosis and relatively low prevalence in the population.
In the present article we set out to give an overview of MODY and describe the importance of considering and confirming the diagnosis of MODY using a family case report.
FAMILY CASE REPORT
Patient 1
Our index patient was diagnosed with gestational diabetes at the age of 31 years. Hyperglycaemia persisted after delivery and five years later a probable diagnosis of type 2 diabetes was made. At that time her body mass index (BMI) was 25 kg/m2 and a C-peptide test showed a good insulin reserve. After initial treatment with diet only, metformin was started but glucose regulation did not improve. A low dose of the sulfonylurea derivative tolbutamide was added and a few years later replaced by insulin therapy. Since the time of diagnosis, diabetic nephropathy (microalbuminuria) and peripheral polyneuropathy developed. Although the patient had received care at our hospital for 25 years and earlier correspondence reported a strong positive family history of diabetes mellitus and cardiovascular disease, no detailed family history was taken. A likely autosomal dominant inheritance pattern of diabetes was discerned in her extended pedigree (figure 1) and genetic testing for the presence of a mutation in the HNF1A gene was performed. The patient (proband IV-8.2 in figure 1) was heterozygous for a variant in HNF1A (c.59G>A, p.Gly20Glu) that had not been detected previously. At the age of 61 her diagnosis was therefore changed to possible HNF1A-MODY. As a consequence, her insulin was stopped and a successful sulfonylurea derivative trial was performed. At the moment her HbA1c is stable at 55 mmol/mol (7.2%) on a diet while taking gliclazide. The patient is currently not using any insulin and reports an increase in the quality of life after stopping insulin therapy. 
Patient 2
The 12 years younger brother of patient 1 (subject IV-8.7 in figure 1) was diagnosed with type 2 diabetes at the age of 24 years based on obesity and preserved insulin reserve during a C-peptide test, after which therapy with NPH insulin once daily was initiated. Major complications of the diabetes developed partly due to the patient’s long withdrawal from care. The patient developed diabetic retinopathy and nephropathy before the age of 40, which progressed to end-stage renal disease, followed by bilateral Charcot feet. At age 44, intensive insulin therapy was started, while three years later gastric bypass surgery was performed because of persistent obesity (BMI approximately 40 kg/m2 ). After his sister’s diagnosis of HNF1A-MODY, the same HNF1A mutation was found to be present and the insulin was withdrawn. Trials with sulfonylurea derivatives and meglitinides (due to postprandial hypoglycaemia with a low-dose sulfonylurea derivative) were performed. Currently the patient is not using any oral glucose-lowering medication or insulin and has stable glycaemic control at 42 mmol/mol on a diet.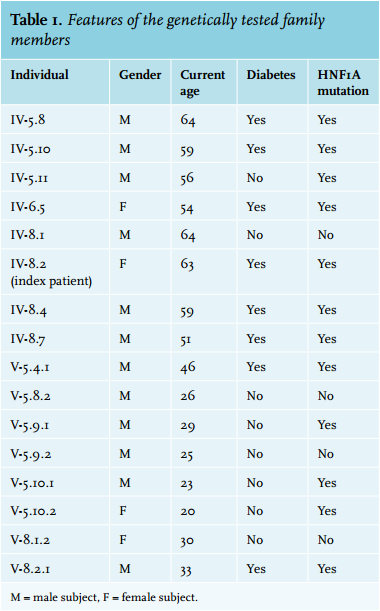
Family
The patients described here came from a large family of Dutch and part Indonesian ancestry. After the probable diagnosis of HNF1A-MODY in the two patients described above, multiple family members with early onset diabetes mellitus came forward for genetic testing. Figure 1 shows the extensive pedigree of the family with the high prevalence of early diabetes and cardiovascular morbidity and mortality and a suggestive autosomal dominant inheritance pattern. Microvascular complications were present in all the known HNF1A-MODY patients in this family, except for the recently diagnosed son of the index patient (V-8.2.1 in figure 1). The age at diagnosis of diabetes mellitus in the family was between 21 and 40 years, with one exception, who was only diagnosed after the discovery of coronary artery disease at the age of 49. All patients from the family with a prior diagnosis of type 2 diabetes were found to be heterozygous for the earlier described variant in HNF1A (table 1). Subsequently, the diagnosis in these patients was changed to HNF1A-MODY and the treating physicians were informed including advice on treatment
In our pedigree we have 12 informative meioses in which the mutation as well as the disease segregate together. A ‘rule of thumb’ is that each meiosis in which the mutation segregates with the phenotype adds 0.3 to the total likelihood of odds (LOD) score, giving a total LOD score in our pedigree of ~3.6: > 3 is a significant association; we could add some additional informative meioses higher up in the pedigree and come to an estimated LOD score of ~5.4. The mutation carriers who have no symptoms (yet) fit in the reduced penetrance that is described for MODY (proband IV-5.11)3 in combination with the relatively young age of some family members (V-5.9.1, V-5.10.1 and V-5.10.2) who may still develop diabetes in the coming years and were advised to undergo yearly check-ups.
EPIDEMIOLOGY
The prevalence of MODY remains unknown but is estimated to be responsible for 1-5% of cases of diabetes mellitus.4-6 As MODY shares clinical features with the more common forms of diabetes mellitus, the true prevalence is probably underestimated.
At present, mutations in 13 genes linked to different types of MODY have been identified.7-9 In table 2 the currently known subtypes of MODY are described with their related proteins/genes and estimated prevalence.10-14
In general, GCK-MODY and HNF1A-MODY each represent 20-70% of all cases, HNF4A-MODY and HNF1B-MODY each account for about 5%, while the other forms are extremely rare.14 GCK-MODY is more commonly diagnosed in countries where glucose testing of asymptomatic people and paediatric cases is routine (Czech Republic, France, Italy, Spain), whereas HNF1A-MODY is more often diagnosed in countries where random blood glucose tests are seldom done and elevated blood glucose is first found after childhood (Denmark, the Netherlands, Norway, United Kingdom).14,15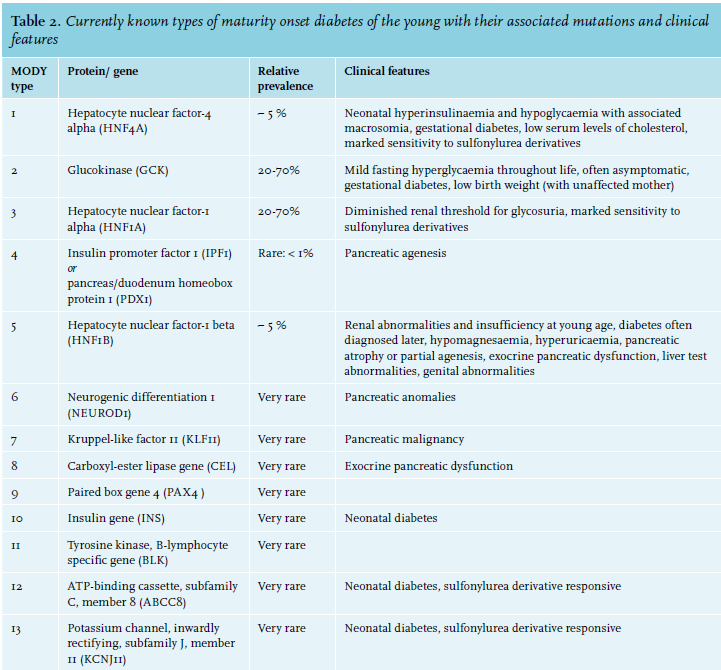
PATHOPHYSIOLOGY
MODY is caused by mutations resulting in pancreatic β-cell dysfunction in the production or excretion of insulin.
Normally, glucose from the circulation is taken up by the β-cell through the glucose transporter type 2 (GLUT 2) on the cell membrane. The enzyme glucokinase (GCK) converts glucose into glucose-6-phosphate which then undergoes glycolysis in the mitochondria to produce adenosine triphosphate (ATP). The increase of the ATP-ADP ratio causes the closure of the β-cell ATP-sensitive potassium channel (KATP), preventing potassium efflux, which leads to depolarisation of the membrane and subsequently causes the opening of the voltage-dependent calcium channels. The subsequent influx of calcium into the cell stimulates exocytosis of insulin-containing granules from the β-cell (figure 2).16
GCK-MODY can be described as disturbed β-cell glucose sensing. Mutations in the GCK gene cause a decrease in glucose metabolism in the β-cell and therefore a rightward shift of the dose-response curve of insulin secretion.
The hepatocyte nuclear factors (HNF)-4 alpha, -1 alpha and -1 beta are transcription factors that form part of a network of transcription factors that controls gene expression of the insulin gene and genes encoding proteins involved in glucose transport and metabolism. Mutations in the genes of these transcription factors lead to reduced expression of these genes in the β-cell and subsequently less insulin production and release. Additionally, HNF-1 beta plays a pivotal role in the development of the kidney, pancreas, liver and genital tract, and mutations subsequently lead to multi-organ consequences.The variant in HNF1A found in the featured family is located in the highly conserved N-terminal HNF-1A dimerisation domain (up to frog considering 11 species). Mutations in this domain disrupt formation of the HNF-1A dimer and the functional DCoH-HNF-1A complex.17 These mutations strongly argue against an obligate dominant negative mode of action and support the idea that glucose homeostasis in humans is sensitive to the dose of HNF-1A. Various in silico algorithms (prediction programs) indicate that the p.Gly20Glu variant is probably damaging. Two other pathogenic amino acid substitutions in the same codon (p.Gly20Arg and p.Gly20Ala)18,19 and 25 other different amino acid substitutions in the dimerisation domain have been described in patients with MODY 3.20 Additionally, the mutation was classified as likely pathogenic based on the American College of Medical Genetics Standards and Guidelines recently published by Richards et al.21
The rarer forms of MODY are caused by mutations in other nuclear transcription factors regulating pancreatic development and expression of the insulin gene, the insulin gene itself (INS-MODY), or genes of proteins involved in the ATP-sensitive potassium channel (ABCC8-MODY and KCNJ11-MODY). They also result in impaired β-cell function with reduced insulin production and/or secretion. 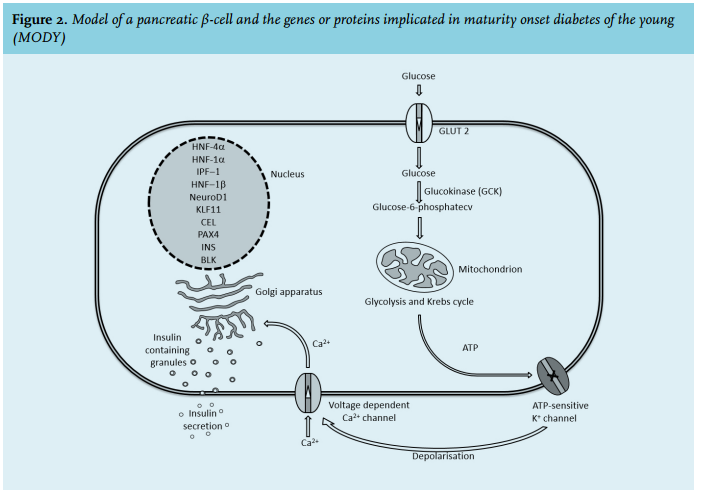
CLINICAL FEATURES
The mode of inheritance of MODY is autosomal dominant. In general, the main characteristics of MODY are the onset of diabetes at a young age with a prominent family history of diabetes in multiple generations and, but not necessarily, absence of obesity,22 as was shown in our HNF1A-MODY family. Patients with MODY may easily be misdiagnosed during pregnancy as gestational diabetes in the context of increased insulin resistance, as described in our index patient. It has been shown that carriers of GCK and HNF1A mutations have an increased risk of gestational diabetes mellitus.22,23
An overview of the typical clinical features of the MODY subtypes is shown in table 2. 7-14,16,22,24-37
TREATMENT
Patients with HNF4A- and HNF1A-MODY are very sensitive to the effect of sulfonylurea derivatives and meglitinides.12,25,28,30,36,38 This efficacy can be explained by the binding with the sulfonylurea receptor type 1 (SUR1) subunit of the KATP causing it to close and triggering opening of the voltage-dependent calcium channels, stimulating insulin release.28,30,36 As meglitinides cause a smaller insulin peak and have a shorter half-life, MODY patients using a meglitinide are less susceptible to hypoglycaemia than during treatment with sulfonylurea derivatives.30,36 Despite the efficacy of sulfonylurea derivative and meglitinide treatment most of these patients will in time require insulin as β-cell dysfunction progresses.14
GCK-MODY, which is often asymptomatic and therefore diagnosed by screening, does not require treatment due to the mild and stable hyperglycaemia with a raised homeostatic set point (glycated haemoglobin (HbA1c) without treatment usually below 64 mmol/mol (8%)).39
It has been shown that HNF1B patients do not respond well to sulfonylurea derivatives and that they have a reduction in insulin sensitivity compared with HNF1A patients.40 If HNF1B patients are treated for their diabetes, the existing studies show that they generally receive insulin.
COMPLICATIONS
Although patients with GCK-MODY are exposed to a lifetime of, albeit limited, hyperglycaemia, microvascular and macrovascular complications in GCK-MODY seem to be limited.41 In patients with HNF1A-MODY both microvascular and macrovascular complications are very common, as was the case in our family. The prevalence is similar to that of patients with type 1 and 2 diabetes, when matched for duration and glycaemic control,42,43 while early detection and treatment may result in a reduced incidence of diabetic complications.44 It has been suggested that the full spectrum of diabetes complications may also occur in HNF4A-MODY patients, particularly retinopathy and nephropathy.10 For HNF1B-MODY it is suggested that microvascular diabetic complications are rare.11,29,34
CASE FINDING
Diagnosing MODY is challenging because of the relatively low prevalence in the general population (1-5%) and shared features with other types of diabetes (table 3). Limited awareness of MODY as a separate entity and cause of diabetes outside specialist centres combined with limited time allocated to enquiring about family history further hampers correct diagnosis, family screening and treatment. A correct diagnosis of MODY might change the treatment to oral glucose-lowering medication (HNF4A-and HNF1A-MODY) or even withdrawal of medication (GCK-MODY). Secondly, certain subtypes are associated with specific medical problems entailing additional diagnostic tests and screening. Lastly, the first-degree relatives of MODY patients have a 50% probability of the same mutation with, at least in the two most commonly occurring MODY forms, a lifetime risk of > 95% of developing diabetes.45,46 As progression of diabetes is generally slow in MODY patients, early diagnosis and start of appropriate treatment might reduce the risk of diabetic complications as described in a Norwegian family.44 The evidence for such an approach is, however, currently lacking and is debatable in light of the possible financial consequences for subjects with a suggestive genotype, but in the absence of diabetes. In general, two simulation studies show that screening may be cost-effective.47,48
Several suggestions have been published for when to consider genetic testing for MODY. Current European guidelines are highly specific, but have a low sensitivity.49 Correspondingly, widening of the clinical criteria for genetic testing can double the number of MODY diagnoses.50 Other recent literature describes additional criteria and Thanabalasingham et al. and Naylor et al. provided algorithms for the consideration of genetic testing.14,51,52 In 2012 a novel approach to correctly allocate genetic testing in Caucasian patients with an onset of diabetes before the age of 35 was published.53 This clinical prediction rule is available as an online calculator (www. diabetesgenes.org/content/mody-probability-calculator).
Using the optimal cut-offs, it has an improved specificity (94% vs 91%) and especially sensitivity (91% vs 72%) for identifying MODY, compared with the criteria of diagnosis < 25 years and a parent with diabetes. Interestingly, applying the calculator to our patients would have advised against genetic testing. Probable reasons for this might be limited accounting for family history and the importance of BMI in the calculator. In addition, a specific HNF1B score focusing on the extra-diabetic features of the disease has recently been developed and validated to select patients for genetic testing of HNF1B-MODY.37
In table 4 we have combined features and algorithms suggestive of possible MODY from the available literature, providing an optional tool for deciding on genetic testing.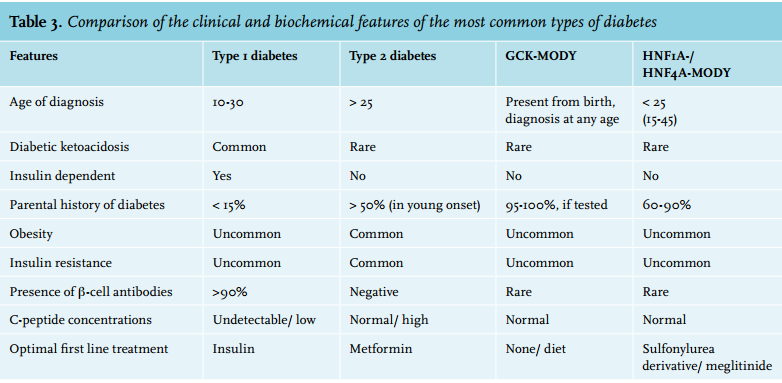

CONCLUSION
Maturity onset diabetes of the young (MODY) encompasses distinct clinical entities causing diabetes. Our presented family illustrates the difficulties in diagnosing MODY, the consequences for treatment with the correct diagnosis as well as the long-term consequences. Correct identification of patients with MODY will probably lead to earlier diagnosis, family screening, earlier and correct treatment and hopefully improved prognosis.
Disclosures
The authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES