KEYWORDS
CAR T-cells, CD19, B-cell malignancies
INTRODUCTION
The cornerstone of the treatment of lymphoproliferative malignancies consists of chemotherapy, regularly followed by autologous or allogeneic stem cell transplantation. These treatments are not tumour cell specific and cause many side effects. In the last decade the focus has moved toward so-called ‘targeted therapies’ using monoclonal antibodies and/or small molecules. Important examples in the current therapeutic arsenal are monoclonal antibodies against CD20, CD52 and CD30, cell surface antigens frequently, but not exclusively, expressed by lymphoproliferative malignancies such as lymphoma and lymphatic leukaemia. Other examples include small molecules targeting intracellular signalling pathways important for growth and survival of malignant cells, such as JAK inhibitors, BTK inhibitors, PI3K inhibitors and inhibitors of the proteasome. Most recently, an effective inhibitor of BCL-2, which induces apoptosis of malignant cells, has become available.1
Another area that is attracting a lot of attention concerns autologous anti-tumour immune responses, in particular by T-lymphocytes. Most tumours are immunogenic but anti-tumour T-cell responses are efficiently suppressed by the tumour through membrane expression of immunosuppressive proteins such as PD-1 ligand and CTLA4 ligand. Interaction of these proteins with PD-1 and CTLA-4 on tumour specific T-cells renders these T-cells anergic. Antibodies that target PD-1 and CTLA-4 interactions (e.g. nivolumab, ipilimumab), release the breaks on the anti-tumour immune response and have been proven very effective.2 Another example is blinatumomab, a bispecific T-cell engager (BiTe). This fusion protein consists of a CD3-specific part (recognising T-cells) and a CD19 specific part (recognising B-cells) and can thereby redirect T-cells directly towards malignant B-cells. Blinatumomab is approved for refractory or relapsed B-cell acute lymphocytic leukaemia (B-ALL).3 Even more sophisticated is the use of engineered CAR T-cells, the topic of this review.
ENGINEERED T-CELL THERAPY IN HAEMATOLOGY
This completely new field involves the administration of autologous T-cells that have been engineered ex vivo to recognise tumour cells. Roughly, there are two ways to employ T-cells against haematological malignancies: T-cell receptor (TCR) engineered T-cells or chimeric antigen receptor (CAR) T-cells (figure 1). TCR T-cells are autologous T-cells transferred with a gene for the α and β chain of a specific T-cell receptor in order to direct the cells towards tumour-associated antigens (for example gp100 and MART-1), cancer germline antigens (for example NY-ESO-1 and MAGE) or cancer-specific mutations (for example in erbb2). TCR T-cells require a specific human leukocyte antigen (HLA) molecule for recognition of the target antigen. Therefore not all individuals are eligible for this treatment. Today, no results from clinical trials using TCR T-cells in haematological malignancies are known; however, several studies targeting the leukaemia antigen Wilms Tumor-1 (WT-1) are on-going. The great advantage of CAR T-cells is that tumour cell recognition can occur without MHC/HLA restriction. Evidence of clinical efficacy in lymphoproliferative malignancies is piling up quickly.4-6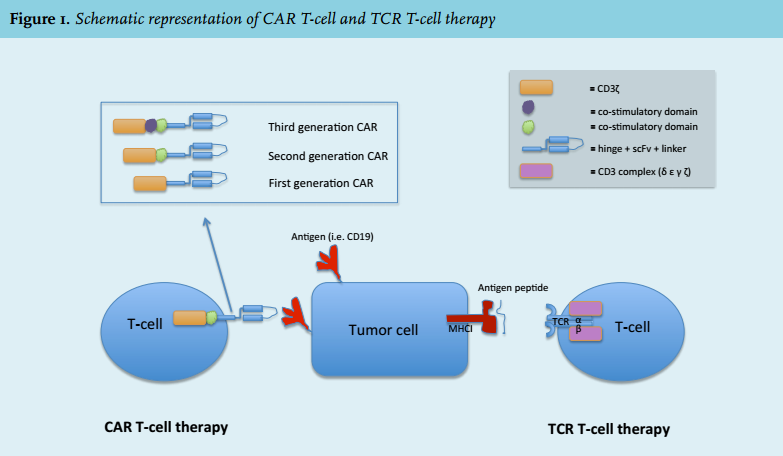
CAR T-CELLS
CAR T-cells are characterised by four important elements (figure 1):
1. A single chain variable fragment (scFv) derived from a murine or humanised antibody which functions as the targeting domain directed against the target of choice. The scFv is attached to an extracellular spacer domain to ensure optimal binding freedom;
2. A hinge and transmembrane domain (often derived from CD8 α or CD28);
3. An intracellular signalling domain that is derived from the TCR (CD3 ζ chain) ensuring T-cell activation;
4. One or two co-stimulatory domains derived from CD28 and/or 4-1BB/CD137. These co-stimulatory domains have been included in the second- and third-generation CAR T-cells, respectively, in order to improve activation, replication and persistence of the CAR T-cells in vivo.
Most of the research to date has focused on autologous CAR T-cells targeting CD19. CD19 is a transmembrane glycoprotein which is expressed on cells of the B-cell lineage, from early pro-B-cells in the bone marrow to mature B-cells in blood and tissues, but not on haematopoietic stem cells. Since CD19 is also expressed on the majority of malignant B-cells, the therapeutic potential of CD19-CAR T-cells is broad.7,8
GENERATION OF CAR T-CELLS
The first step in the generation of autologous CAR T-cells is the collection of peripheral blood mononuclear cells through leukapheresis. The cells are cultured in the presence of a T-cell specific mitogenic stimulus, often using beads coated with anti-CD3/anti-CD28 monoclonal antibodies, resulting in the activation of T-cells with high replicative capacity.7,8 The second step is transduction or transfection of the CAR construct, which is directed against the target antigen of choice. Multiple methods have been tried including transposon-based systems introducing plasmid DNA into T-cells or electroporation of mRNA into T-cells, but retroviral or lentiviral transduction is currently the transduction mode of choice. CAR T-cells are then cultured in the presence of cytokines (i.e. IL-2, IL-7, IL-12, IL-15). After expansion for several days, the cells are re-infused into the patient. In almost all CD19 CAR T-cell studies patients are lymphodepleted before infusing the engineered T-cells. In B-ALL patients the conditioning therapy mostly consists of fludarabine and cyclophosphamide, in lymphoma patients the choice depends on previous individual treatments. Pre-infusion lymphodepletion facilitates expansion and persistence of CAR T-cells in vivo.7-10
CLINICAL USE OF CD19 CAR T-CELLS
After preclinical evaluation, CD19 CAR T-cells were first tested in children and adults with relapsed/refractory ALL in adults with advanced relapsed and refractory chronic lymphatic leukaemia (CLL). Later patients with B-cell non-Hodgkin lymphomas were included as well. Table 1 demonstrates the phase 1-2 trials which have been performed and published so far, each including ten or more patients.11-23 Several smaller trials have been performed as well, which are reviewed in other papers.24,25 Taken together, clear and deep responses are seen in a substantial percentage of B-ALL patients (complete responses in 70-94% of the cases). Durable complete responses correlate with persistence of the CAR T-cells and lasting B-cell depletion. It remains to be determined to what extent CD19 CAR-T cells may be curative for B-ALL and whether it has the potential to replace allogeneic stem cell transplantation in the future. More detailed studies will shed light on the causes of the disappearance and exhaustion of CAR T-cells that heralds B-ALL relapse.13-15,18,21,23-25
Data on patients with diffuse large cell B-cell lymphomas are still scarce but so far complete response rates range from 20-100%.19,20,22,25
Strikingly, patients with CLL appear to have lower response rates in the generally small trials performed (complete responses in 20-75% of the patients).16,17,19,22,25 Although not fully elucidated yet, this might be due to disease-specific properties of CLL, in particular the interaction of CLL cells with the tumour microenvironment, which may cause exhaustion and dysfunction of (CAR) T-cells.26
In other types of B-cell lymphomas (mantle cell lymphoma, follicular lymphoma) still only limited data are available.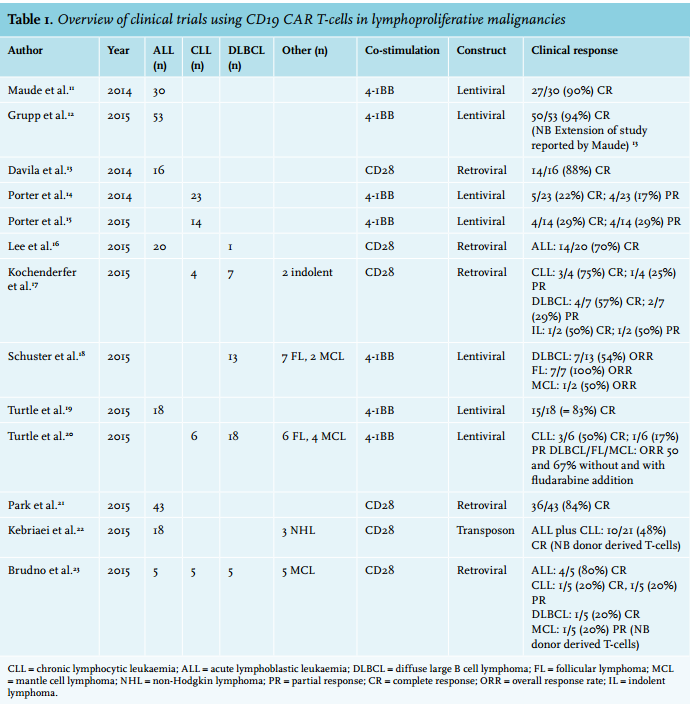
THE DOWNSIDES OF CD19 CAR T-CELLS
There are several serious side effects of CAR T-cell therapy, which are mostly on-target toxicities. CD19 CAR T-cells specifically target CD19 positive B-cells, including healthy B-cells, resulting in long lasting B-cell aplasia. This aplasia – a surrogate for the persistence of CAR T-cells – puts successfully treated patients at risk for infections. Intravenous immunoglobulin therapy can prevent the majority of these infections. In addition, many patients develop cytokine release syndrome with symptoms ranging from mild and flu-like symptoms to severe reactions and multi-organ failure. The latter occurs in 30% of patients, typically 5-21 days after the infusion of CAR T-cells and is mediated by elevated levels of circulating cytokines, especially IL-6, IL-10 and interferon gamma. Treatment with the IL-6 inhibitor tocilizumab is highly effective; however, it is unclear whether this treatment affects the anti-tumour response. The same holds true for treatment with steroids. Finally, a number of patients have developed neurological symptoms ranging from seizures to aphasia and delirium. This type of toxicity seems transient and its pathophysiology is ill-understood.7,8
FUTURE DIRECTIONS OF CART - CELL THERAPY
Current research focuses on improving the safety and efficacy of CAR T-cell therapy. To be able to switch off the CAR T-cell response, for example in case of excessive toxicity, ‘suicide genes’ have been incorporated into the T-cells. Examples are introduction of thymidine kinase, which can be inhibited by ganciclovir, expression of inducible caspase or Fas (pro-apoptotic genes) or expression of surface proteins rendering CAR T-cells susceptible to existing agents such as rituximab or cetuximab. Making CAR expression inducible instead of permanent could also increase safety.4,27
To increase efficacy, multiple approaches are being explored. Firstly, it is investigated whether specific T-cell subsets provide superior antitumour efficacy. Currently, αβ T-cells (expressing a TCR consisting of an α and β chain) are used; however, therapy with γδ T-cells, specifically the Vγ9Vδ2 subset, may be more effective as these cells specifically recognise and efficiently kill cells with increased levels of phosphoantigens, which have been found in the majority of B-cell lymphomas.28 Furthermore, it has become clear that selecting less differentiated T-cells such as naïve T-cells, T-memory stem cells and central memory T-cells for transduction results in more effective in vivo expansion and survival.4 To render CAR T-cells less susceptible to the suppressing effects of regulatory T-cells, T-cell signalling has been altered by increasing the expression of AKT or by modifying the CD28 signalling domain.28 Interestingly, such an approach could also be of value in improving CAR T-cell therapy in CLL, as T-cells are globally dysfunctional in CLL with a few notable exceptions that could be enforced.29
A second approach to improve efficacy is to develop CAR T-cells that express transgenes for IL-2, IL-12, IL-15 or IL-21. These cytokines are important for immune stimulation and may ‘armour’ the infused CAR T-cells. In addition, chemokine receptors such as IL-7Rα have been built in to improve CAR T-cell survival. Thirdly, dual or polyspecific CAR T-cells might create higher tumour specificity. An example is the development of CAR T-cells targeting both CD19 and CD5 for CLL.27 Furthermore, combination therapy with, for example, immune checkpoint inhibitors (inhibiting PD-1 and CTLA4) is being piloted which has the potential to overcome inhibition of the CAR T-cell response.27,30 To make CAR T-cells more readily available (‘off the shelf’) the use of allogeneic CAR T-cells is being explored. This obviously poses additional risks of graft-versus-hostdisease, which can be overcome by knockdown of the autologous T-cell receptor or introduction of a suicide gene.29
In the near future, several clinical trials with CAR T-cells will also become available in the Netherlands (for more information see www.hovon.nl/werkgroepen/llpc).
CONCLUSION
CAR T-cells provide a promising new type of treatment for lymphoproliferative malignancies. Clinical trials in patients with relapsed or refractory B-ALL and different types of B-cell lymphomas demonstrate impressive and often durable responses. The current clinical experience in haematology thus far mostly involves the use of CAR T-cells targeting the B-cell antigen CD19. Developments in novel and more specific target identification, dual target approaches, T-cell subset selection and CAR T-cell engineering will make this treatment even more safe, specific and effective. Hopefully, these developments will bring adoptive T-cell therapies further forward with the potential to replace stem cell transplantation in patients with lymphoproliferative malignancies.
DISCLOSURES
M.J. Kersten and A. Hagenbeek have received financial compensation for advisory boards for Novartis.
REFERENCES