What was known on this topic?
Familial focal segmental glomerulosclerosis (FSGS) is caused by mutations in genes which have an important role in the function of podocytes. Several genes have been identified; however, in many families the cause remains unknown. Research on gene mutations and comprehensive investigation of phenotypes will teach us more about the disease and this will result in early diagnosis, better prediction of prognosis for the individual patient, genetic counselling and hopefully this will contribute to directed personalised therapy in the future.
What does this add?
We present a family with a mutation in the gene encoding inverted formin 2 (INF2), a known cause of familial FSGS. However, the initial suspected diagnosis was Alport syndrome. This case report creates awareness of the large variability in phenotypic expression of FSGS caused by a mutation in one of the genes affecting the podocyte. Namely, familial FSGS can present with nephrotic or non-nephrotic range proteinuria, with or without microscopic haematuria, and with or without neurological symptoms. Awareness of the variability in presentation of FSGS caused by a genetic defect in combination with a thorough family history taking is important to diagnose these patients and emphasise the importance of genetic testing. Moreover, this is the first report with a comprehensive phenotypic description of a family with a mutation in INF2 and a short overview of the knowledge about other patients described in literature with a mutation in the INF2 gene.
INTRODUCTION
Focal segmental glomerulosclerosis (FSGS) is one of the most common patterns of glomerular injury. FSGS is a very heterogeneous disease with many underlying genetic and environmental causes. Here we report on a family who presented with haematuria and proteinuria, clinically resembling Alport syndrome. Further analysis revealed FSGS due to a mutation in the gene encoding inverted formin 2 (INF2).
CASE REPORT
Patient 1, a 31-year-old Caucasian female, presented with oedema of the lower extremities. Additional work-up revealed microscopic haematuria and proteinuria of 2.7 g/24 hours, a normal serum albumin (44 g/l) and renal insufficiency (endogenous creatinine clearance 40 ml/ min). Based on renal biopsy, a diagnosis of focal segmental extracapillary glomerulonephritis was established. The patient did not receive any immunosuppressive drugs. Within three years after diagnosis she progressed to end-stage kidney disease (ESKD) and haemodialysis was started. One year later, a postmortal kidney transplantation was performed and the graft functioned well until the patient died at the age of 64. The family history revealed that the patient’s mother died at the age of 40 due to an unknown renal disease. Her brother had developed ESKD at the age of 26 and was treated with haemodialysis.
Patient 2, the son of patient 1, tested negative for proteinuria and haematuria at the age of 10. However, at the age of 18, he was referred to the outpatient clinic because of microscopic haematuria and asymptomatic proteinuria (5 g/24 hours). Serum albumin (38 g/l) and renal function (endogenous creatinine clearance 115 ml/min) were normal. Physical examination, including neurological examination, was unremarkable. The renal biopsy report described ten glomeruli of which two with complete sclerosis. Open glomeruli showed increased mesangial matrix, focal reduplication of capillary loops, synechiae and sclerosis. Immunofluorescence showed depositions of IgM along the capillary wall and within the mesangium. Immunofluorescence for type IV collagen was not performed. Electron microscopic examination was described as sporadic splitting of the lamina densa with augmented mesangial matrix. Based on the renal biopsy, the microscopic haematuria and the pattern of inheritance, an X-linked inherited disease (Alport syndrome) was suspected. However, an audiogram was negative for hearing loss at age 23 years and Sanger sequencing showed no mutation in the COL4A5 gene. Five years after presentation, ESKD developed. Two years later a postmortal kidney transplantation was performed. After 23 years, the graft is still functioning well.
Patient 3, the son of patient 2, was referred to our outpatient clinic at the age of 17 because of asymptomatic proteinuria (1.8 g/24 hours) without haematuria. His past medical history was negative. Physical examination was unremarkable. Serum albumin (44 g/l) and renal function (endogenous creatinine clearance 150 ml/min) were normal.
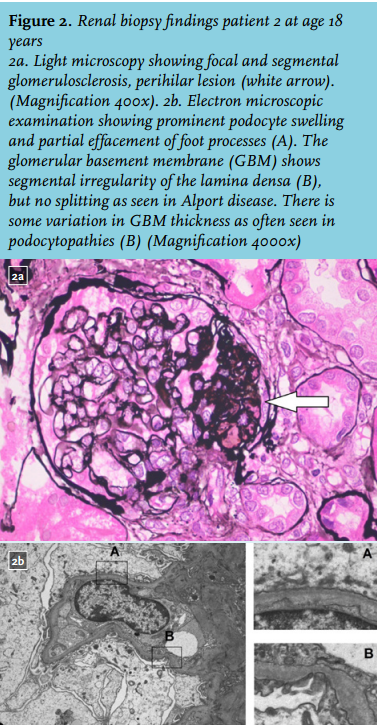
After the referral of patient 3, an X-linked disease was excluded and an autosomal dominant pattern of inheritance was suspected (figure 1). Subsequently, we reviewed the renal biopsy specimens from patient 2; slides from patient 1 were no longer available. Light microscopic examination showed primarily focal segmental sclerotic lesions (perihilar and tip lesions; figure 2a), review of the electron microscopic images revealed a sporadic irregular lamina densa with alternating thickening and thinning of portions of the glomerular basement membrane. Because the lesions were sporadic, stored renal biopsy material was used for additional electron microscopic examination. These images all showed partial foot process effacement and some variation in glomerular basement membrane thickness as often seen in podocytopathies (figure 2). Based on this information, a diagnosis of familial FSGS was made. Additional diagnostic work-up compromised genetic screening for mutations causing autosomal dominant FSGS, according the Dutch guideline for FSGS.1,2
Analysis of the inverted formin 2 (INF2) gene revealed a heterozygous missense mutation c.529C>T (p.(Arg177Cys)) within the diaphanous inhibitory domain of the protein. This mutation is not reported in our in-house variant database containing exome sequencing variants detected in 5031 individuals, nor in ExAC, a large database collecting variants in over 60,000 exomes as proxy for variant allele frequencies in the general population.3 The mutation has only been described once in the literature in a patient with FSGS.4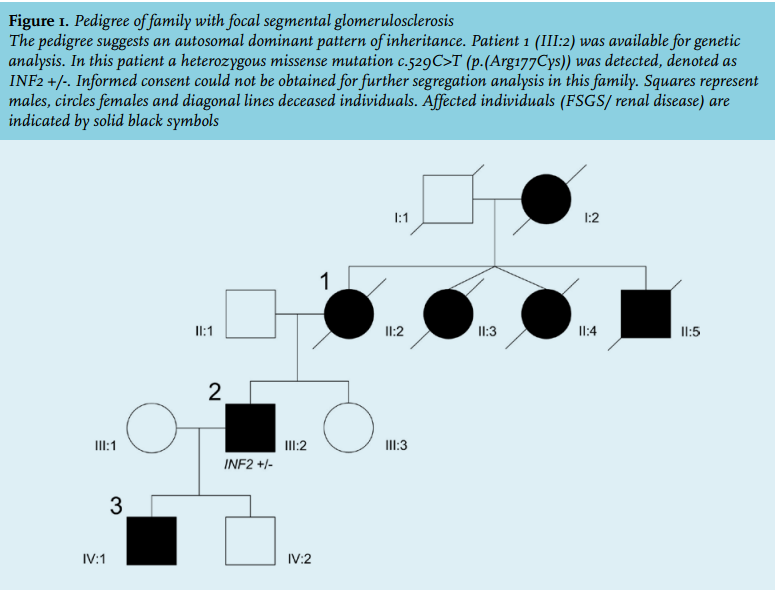
 DISCUSSION
DISCUSSION
Injury to podocytes plays a central role in the pathogenesis of FSGS and the aetiology is diverse, including mutations in genes encoding proteins that are crucial for the function of podocytes and the glomerular basement membrane.5 The prevalence of gene mutations is especially high in children with sporadic steroid-resistant FSGS and in patients with a family history of FSGS.1 Genetic testing should be considered in these patients, especially because results can affect the care of the patient and their family. In this case report we describe a family with a missense mutation in the INF2 gene, initially thought to have Alport syndrome.
In 2010, the INF2 gene was identified as a novel gene causing autosomal dominant FSGS.6 INF2 mutations are currently one of the most common causes of autosomal dominant FSGS (12-17%), but are rarely found in sporadic cases of FSGS.6-8 Until now, up to 44 different mutations in INF2 have been reported.4,7,9,16 Patients with a mutation in the INF2 gene typically present during adolescence or early adulthood with moderate to nephrotic range proteinuria, frequently progressing to ESKD in the third or fourth decade of life. Some patients present with microscopic haematuria or hypertension.6,8 The clinical expression of INF2 mutations is highly variable, even in individuals carrying the same genetic mutation.4,6,8,9,11-13 It is hypothesised that mutations in other (FSGS) genes or environmental factors contribute to this variability.8
INF2 belongs to the formin family, a group of actin-binding proteins that regulate a variety of cytoskeleton-dependent cellular processes. INF2 is widely expressed in podocytes and mutations in the INF2 gene are thought to cause FSGS by dysregulation of the podocyte skeleton.17 INF2 proteins are also present in Schwann cells and play a role in myelination and myelin maintenance. In approximately 75% of patients with Charcot-Marie-Tooth (CMT) disease associated with FSGS, a mutation in INF2 was identified.9 CMT is a disorder affecting peripheral motor and sensory neurons which can cause progressive distal muscle weakness and atrophy, reduced tendon reflexes, sensorineural hearing loss, and deformities in feet and hands.18 However, presenting symptoms can be discrete: some patients have only pes cavus as a clinical sign.9 Therefore a careful clinical neurological examination and screening for hearing loss should be considered in all patients with FSGS, including patients with a negative family history, since de novo INF2 mutations appear to be more common in patients with CMT disease with FSGS in comparison with isolated FSGS.9,14 The reason why INF2 mutations do not always lead to CMT may be related to the location of the mutation. All mutations are located in the diaphanous inhibitory domain of the protein. In patients with FSGS and CMT, INF2 mutations are mainly located in exon 2 and 3. However, there are also patients described in the literature with a mutation in exon 2 or 3 with isolated FSGS. Whether this is a phenotypic variation or lack of thorough phenotypic description is unclear. In contrast, mutations in exons 4 and 6 resulted in isolated FSGS.4,7-14 This information is based on single publications reporting a mutation in INF2. However, there are no studies available with a clear genotype-phenotype correlation.
Initially, Alport syndrome was considered in this family. Alport-like lesions have been described previously in INF2 mutations.12 Lee et al. presented a 9-year-old Korean boy with a heterozygous c.658G>A (p.(Glu220Lys) mutation of the INF2 gene. Renal biopsy revealed FSGS lesions with irregular podocyte foot processes and focal thinning and lamellation of the glomerular basement membrane, mimicking Alport syndrome. However, expression of α3 and α5 chains of type IV collagen was normal in immunofluorescence microscopy.
In conclusion, we present a family with a mutation in INF2 initially thought to have Alport syndrome. Awareness of the heterogeneous phenotypic expression of a mutation in INF2 is important and this case illustrates the importance of the history taking of the family, thorough examination of kidney biopsy, and importantly the combination with a molecular diagnosis.
DISCLOSURES
IMR is supported by an AGIKO grant from ZonMw-NWO (grant 92003587).
The authors declare no conflicts of interest.
REFERENCES