KEYWORDS
Acromegaly, treatment, somatostatin analogues, cabergoline, pegvisomant, pasireotide, oral octreotide
INTRODUCTION
Acromegaly is a rare disease characterised by excessive growth hormone (GH) secretion almost exclusively caused by a benign pituitary adenoma.1 Clinical features are the result of chronic GH and insulin-like growth factor-1 (IGF-I) hypersecretion leading to soft tissue enlargement, excessive skeletal growth, metabolic disturbances, a reduced life expectancy and a reduced quality of life (QoL).1,2. Incidence of acromegaly is estimated to be around 2.8-6 cases per million per year.3-8 However, this is an underestimation because many cases go unrecognised, as data from a detailed population-based study in Belgium reported that the true incidence of acromegaly might be 1 case per 8000 population, which suggests that acromegaly is more prevalent than previously considered.9,10 Many signs and symptoms develop insidiously and are often subtle, particularly in the early stages before the characteristic physical changes become visible. Historically, the treatment delay from first symptoms to diagnosis is 7-10 years, although in younger patients the delay seems to be shorter.11
Optimal management of acromegaly is based on three pillars: control of GH and IGF-I hypersecretion, tumour size control and optimisation of QoL by comprehensive management of the comorbidities commonly associated with acromegaly, such as diabetes mellitus, hypertension, obstructive sleep apnoea and dyslipidaemia. Surgical, medical and radiotherapy modalities are available to treat acromegaly. An optimal treatment approach should be chosen depending on the size, localisation of the pituitary adenoma and patient characteristics.
This article will review the current therapies with a focus on the recent significant advances in the medical treatment of acromegaly.
DIAGNOSIS AND ASSAY PITFALLS
The clinical manifestations of acromegaly depend on the progression of the disease and patients may not always manifest with clear diagnostic features. Clinicians should be aware of the possibility of acromegaly in patients with two or more of the following comorbidities: new-onset diabetes, diffuse arthralgias, new-onset or difficult-to-control hypertension, cardiac disease including biventricular hypertrophy and diastolic or systolic dysfunction, fatigue, headaches, carpal tunnel syndrome, sleep apnoea syndrome, diaphoresis and loss of vision.12
The biochemical diagnosis of acromegaly is made by measurement of serum IGF-I which, because its half-life is longer than that of GH, serves as an integrative marker of GH secretion. Another advantage of a single IGF-I over a single GH measurement is that IGF-I can be assessed independent of the time of day and food intake. In cases that are not clear-cut, i.e. with IGF-I levels just above or around the upper limit of normal, confirmation is frequently needed by showing a lack of suppression of GH to less than 1 μg/l following documented hyperglycaemia during an oral glucose tolerance test.12 In acromegaly patients with poorly controlled diabetes mellitus, the oral glucose tolerance test is not reliable and serum IGF-I levels should be re-assessed when glycaemic control has been established. Systemic illnesses, hepatic or renal failure, malnutrition, diabetes mellitus and oral oestrogens may decrease IGF-I levels which might result in false-negative interpretations.12-14 False-positive elevated IGF-I levels can occur during pregnancy. Accurate measurement of GH and IGF-I is important for diagnosis and monitoring of acromegaly. In an illustrative paper, the same GH sample was measured in 104 centres and an IGF-I sample in 23 centres across the UK using different assays. The results varied more than threefold for GH and about 2.5 fold for IGF-I.15 Even when using the same automated immunoassay, significant intraindividual variability still existed. It is, therefore, imperative that when assessing serum IGF-I levels, the values should be interpreted against the clinical background rather than by the absolute IGF-I values alone. An overview of the current treatment modalities is shown in table 1.
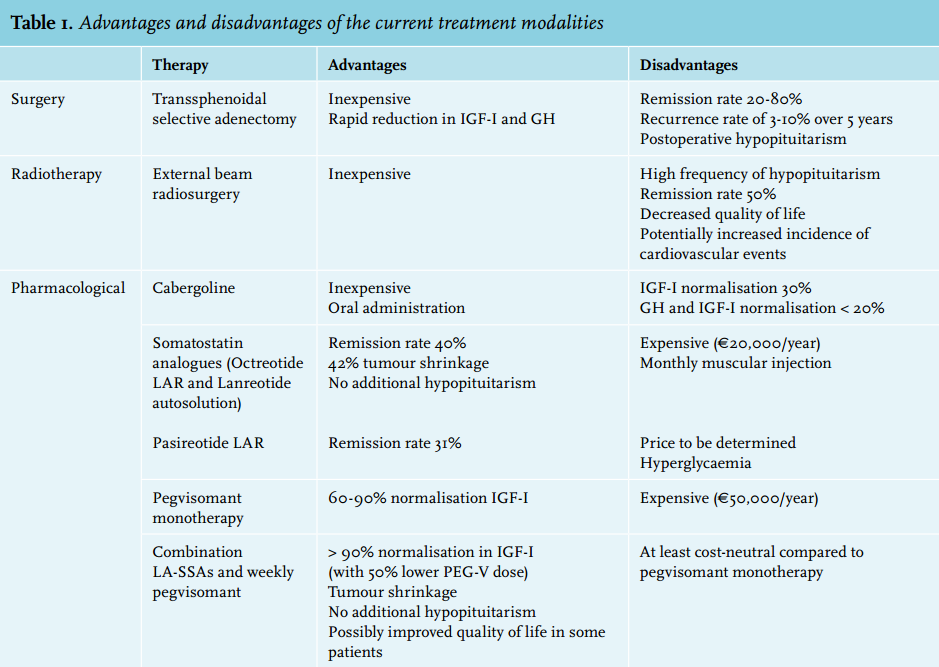
Surgery
Transsphenoidal surgery is the primary treatment for patients with small and, therefore, curable tumours or for large adenomas causing impingement of the optic chiasm.16,17 Surgical results depend on preoperative GH and IGF-I levels, tumour invasiveness and surgical skills. Surgery is the treatment of choice for microadenomas (diameter ≤ 1 cm) and well-defined intrasellar macroadenomas. In these cases experienced surgeons can achieve remission rates of about 80%, defined as postoperative normalisation of IGF-I levels and suppression of GH levels to < 1 mg/l after an oral glucose load. These rates drop to 20-30% for macroadenomas (diameter ≥ 1 cm). For macroadenomas that are not entirely surgically resectable (e.g., those with cavernous sinus extension) surgery may be considered with the goal of debulking the tumour mass.
Debulking seems to increase the efficacy of postoperative treatment with long-acting somatostatin analogues (LA-SSAs),18-20 although conflicting data have been reported as well.21 Preoperative treatment of macroadenomas with LA-SSAs has been shown to improve surgical outcome.22-25 Successful surgery is accompanied by a rapid fall in GH and IGF-I levels and the costs are relatively low compared with life-long drug therapy, although the efficacy of surgery might be overestimated as the data on surgical outcome are almost exclusively reported from high specialist centres. For instance, in the UK, for all centres, an efficacy rate for microadenomas < 37% and for macroadenomas < 20% has been reported.26
Radiotherapy
Although radiotherapy has been used for decades, nowadays it is considered a third line of treatment for acromegaly in most centres.27-29 For conventional radiotherapy, remission rates of around 50% after a follow-up of ten years have been reported. However, these remission rates are accompanied by an increased risk of hypopituitarism as 50-80% of patients develop pituitary insufficiencies after a mean follow-up period of ten years.30,31 Another drawback is that it sometimes takes years before radiotherapy induces biochemical remission, which is associated with a negative impact on quality of life.32 Analysis of the UK acromegaly database showed that radiotherapy was associated with an increased mortality risk, and cerebrovascular disease as the main cause of death.33 Additionally, studies assessing QoL observed a lower QoL in patients treated with radiotherapy that further decreased during follow-up.32,34,35 Joint problems are important factors affecting the QoL after radiotherapy.34 In patients in whom an increase in tumour size is observed despite surgery and medical therapy, radiotherapy should be considered. Very rarely, pituitary adenomas still increase in size after radiotherapy.
Pharmacotherapy
Somatostatin analogues
Somatotroph (i.e. GH secreting) adenomas predominantly express somatostatin receptor sub-type 2 (SSTR2) and 5 (SSTR5). Octreotide long-acting release (Sandostatin LAR™) and lanreotide (Somatulin autosolution™) are long-acting analogues of somatostatin (growth hormone inhibitory hormone) that inhibit GH secretion by predominantly binding to SSTR2. Both formulations are on the market as monthly injections and equivalent in terms of efficacy, but differ in their mode of administration; lanreotide is available in pre-filled syringes injected deep subcutaneously and octreotide LAR requires reconstitution before being injected intramuscularly.36,37 Biochemical normalisation of IGF-I and GH levels can be obtained in about 40% of treatment-naïve patients with LA-SSAs.38,39 Tumour shrinkage is frequently observed (40-63%) during LA-SSA treatment and the decrease in GH levels generally occurs within the first four months.39-43 LA-SSAs have a good safety and tolerability profile. Relatively few side effects occur; in the first few weeks transient self-limiting gastrointestinal symptoms such as abdominal discomfort, nausea and fat malabsorption occur in most patients.44 Asymptomatic gallbladder stones or bladder sludge can develop in the first 18 months in up to 20% of patients.45 Although current guidelines do not yet recommend the use of preoperative LA-SSAs, there is clear evidence from a meta-analysis and three randomised controlled trials indicating that LA-SSAs can improve the efficacy of surgery in macroadenomas.46,47 However, there is limited evidence that LA-SSAs improve surgical outcome. LA-SSA pretreatment is a good option for patients with macroadenomas and for patients on a waiting list for neurosurgery, as it can reduce signs and symptoms.
Dopamine agonists
Until the 1980s, dopamine agonists were the only class of pharmaceutical agents available for acromegaly. Cabergoline is an oral second-generation dopamine agonist with a high affinity for dopamine receptor type 2 and has been used as monotherapy and in combination with somatostatin analogues.48 It is usually well tolerated with few side effects and is inexpensive.49 Because cabergoline alone has a modest efficacy of about 30% in normalising IGF-I levels, it is recommended as an add-on therapy in patients who have not reached biochemical remission on somatostatin analogues alone, and for patients with no access to pegvisomant.48,50 However, the efficacy of cabergoline to control IGF-I and GH is probably below 20%.48
Pegvisomant
Pegvisomant (Somavert®) is a genetically modified analogue of human GH that binds to and blocks the GH receptor, acting as a competitive growth hormone receptor antagonist.51 It is currently used as a second-line therapy in patients who are inadequately controlled with LA-SSA monotherapy.27 Treatment with pegvisomant results in a rapid reduction in IGF-I serum levels which causes a paradoxical rise in serum GH levels, due to the negative feedback loop via the hypothalamus and the pituitary gland.52,53 Cross-reactivity between pegvisomant and endogenous GH in commercial assays disables proper assessment of the endogenous GH levels.54 For these reasons GH cannot be reliably assessed in patients treated with pegvisomant, unless specific assays are used.55 The key biomarker during the treatment of pegvisomant, therefore, is the serum IGF-I level along with clinical signs and symptoms.
To date, pegvisomant is the most effective drug to normalise IGF-I levels in acromegaly.51,53,56 Reports from clinical studies demonstrated that more than 90% of patients with acromegaly achieved normalised IGF-I levels.53,56 Because pegvisomant is a competitive blocker, virtually all patients with acromegaly can be controlled providing treating physicians adequately titrate the dose of pegvisomant. However, in observational registries lower efficacy rates of around 60% were reported.57-60 The lower efficacy might be explained by the relatively low doses of pegvisomant that were recorded in these registries. To achieve efficacy rates of above 90% with pegvisomant monotherapy, the average expected weekly dose is probably above 120 mg. Efficacy rates of pegvisomant as a single agent and in combination with LA-SSAs are equally high. However, the advantage of co-administration of pegvisomant with LA-SSAs is the much lower (around 50%) required weekly dose of pegvisomant.52 Because LA-SSAs inhibit the secretion of GH, pegvisomant meets less competition from endogenous GH around the GH receptor, meaning a lower dose of pegvisomant is needed to block all GH receptors during combination therapy and additionally reduces the number of GH receptors on the hepatocytes.52,61,62 Although LA-SSA treatment decreases hepatic IGF-I production, GH action in peripheral tissues remains too high. This may lead to insufficient control of disease activity in peripheral tissues despite biochemical control. Blocking peripheral GH action using pegvisomant can therefore be useful in treating extrahepatic acromegaly.63 Improvement in quality of life was previously observed in acromegaly patients who had normalised IGF-I during LA-SSA therapy.64 Pegvisomant therapy has also been shown to have beneficial effects on glucose metabolism by several mechanisms.65-70
Pegvisomant is also an expensive drug. A median dose of 120 mg per week comes at an annual price of around € 62,000. Combining pegvisomant with LA-SSAs, therefore, might significantly reduce medication costs. The most common side effect associated with the use of pegvisomant is a transient elevation of liver transaminases.52,58,60-62 The incidence of this temporary increase in transaminases was reported to be higher in patients on a combination with LA-SSAs. Although many risk factors have been suggested, the underlying pathophysiology of the development of a pegvisomantinduced transient elevation of liver transaminases remains unclear.61,62,71,72 There is no clear evidence that pegvisomant directly promotes tumour growth, but ongoing vigilance is required by repetitive imaging to monitor tumour size.58 The high efficacy of pegvisomant in acromegaly can only be achieved in experienced centres that treat a high volume of patients with pegvisomant.52
Pasireotide
Pegvisomant (Somavert®) is a genetically modified analogue of human GH that binds to and blocks the GH receptor, acting as a competitive growth hormone receptor antagonist.51 It is currently used as a second-line therapy in patients who are inadequately controlled with LA-SSA monotherapy.27 Treatment with pegvisomant results in a rapid reduction in IGF-I serum levels which causes a paradoxical rise in serum GH levels, due to the negative feedback loop via the hypothalamus and the pituitary gland.52,53 Cross-reactivity between pegvisomant and endogenous GH in commercial assays disables proper assessment of the endogenous GH levels.54 For these reasons GH cannot be reliably assessed in patients treated with pegvisomant, unless specific assays are used.55 The key biomarker during the treatment of pegvisomant, therefore, is the serum IGF-I level along with clinical signs and symptoms.
To date, pegvisomant is the most effective drug to normalise IGF-I levels in acromegaly.51,53,56 Reports from clinical studies demonstrated that more than 90% of patients with acromegaly achieved normalised IGF-I levels.53,56 Because pegvisomant is a competitive blocker, virtually all patients with acromegaly can be controlled providing treating physicians adequately titrate the dose of pegvisomant. However, in observational registries lower efficacy rates of around 60% were reported.57-60 The lower efficacy might be explained by the relatively low doses of pegvisomant that were recorded in these registries. To achieve efficacy rates of above 90% with pegvisomant monotherapy, the average expected weekly dose is probably above 120 mg. Efficacy rates of pegvisomant as a single agent and in combination with LA-SSAs are equally high. However, the advantage of co-administration of pegvisomant with LA-SSAs is the much lower (around 50%) required weekly dose of pegvisomant.52 Because LA-SSAs inhibit the secretion of GH, pegvisomant meets less competition from endogenous GH around the GH receptor, meaning a lower dose of pegvisomant is needed to block all GH receptors during combination therapy and additionally reduces the number of GH receptors on the hepatocytes.52,61,62 Although LA-SSA treatment decreases hepatic IGF-I production, GH action in peripheral tissues remains too high. This may lead to insufficient control of disease activity in peripheral tissues despite biochemical control. Blocking peripheral GH action using pegvisomant can therefore be useful in treating extrahepatic acromegaly.63 Improvement in quality of life was previously observed in acromegaly patients who had normalised IGF-I during LA-SSA therapy.64 Pegvisomant therapy has also been shown to have beneficial effects on glucose metabolism by several mechanisms.65-70
Pegvisomant is also an expensive drug. A median dose of 120 mg per week comes at an annual price of around € 62,000. Combining pegvisomant with LA-SSAs, therefore, might significantly reduce medication costs. The most common side effect associated with the use of pegvisomant is a transient elevation of liver transaminases.52,58,60-62 The incidence of this temporary increase in transaminases was reported to be higher in patients on a combination with LA-SSAs. Although many risk factors have been suggested, the underlying pathophysiology of the development of a pegvisomantinduced transient elevation of liver transaminases remains unclear.61,62,71,72 There is no clear evidence that pegvisomant directly promotes tumour growth, but ongoing vigilance is required by repetitive imaging to monitor tumour size.58 The high efficacy of pegvisomant in acromegaly can only be achieved in experienced centres that treat a high volume of patients with pegvisomant.52
Pasireotide
Pasireotide LAR (Signifor®) is a novel multireceptor somatostatin analogue with broader somatostatin receptor binding affinity for SSTR1, SSTR3 and SSTR5. It is available as monthly subcutaneous injections. Because of the broader binding profile compared with octreotide and lanreotide, it may provide additional therapeutic benefits. Pasireotide LAR has recently been approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of acromegaly in patients in whom surgery is not an option or is not curative and who are inadequately controlled on treatment with first-generation somatostatin analogues.73
In a prospective, randomised, double-blind head-to-head superiority study in medically naïve acromegaly patients, biochemical control after 12 months was significantly higher in the pasireotide LAR compared with the octreotide LAR treated patients (31.3% vs. 19.2%). However, hyperglycaemia-related adverse events were more common in the pasireotide LAR treated group (57.3% vs. 21.7%).74 Recently, the efficacy and safety of pasireotide LAR was addressed in acromegaly patients refractory to octreotide LAR or lanreotide autosolution. Inadequately controlled acromegaly patients on the currently available somatostatin analogues were randomised to pasireotide LAR 40 mg, pasireotide LAR 60 mg or continued treatment with octreotide LAR 30 mg or lanreotide autosolution 120 mg (active control). After 24 weeks, biochemical control was achieved in 15% of patients in the pasireotide LAR 40 mg group, 20% in the pasireotide LAR 60 mg group and no patients in the active control group. IGF-I normalisation was reported in about 25% of patients in both pasireotide LAR groups, while no patients receiving active control achieved normal IGF-I concentrations.75 Glycosylated haemoglobin levels increased in the pasireotide group within 12 weeks and remained elevated throughout the study. Because somatostatin receptors are also expressed on pancreatic islet cells, somatostatin analogues may also affect glucose homeostasis. Several studies have demonstrated that pasireotide is associated with a higher frequency and severity of hyperglycaemia. Mechanistic studies in healthy volunteers have suggested that the hyperglycaemic effect of pasireotide is related to decreases in insulin secretion and incretin hormone response, but with no effect on insulin sensitivity. Pasireotide-induced hyperglycaemia can be managed with standard antidiabetic treatment with a possible additional beneficial effect of dipeptidyl peptidase-4 (DPP-4) inhibitors and glucagon-like peptide-1 (GLP-1) agonists.76,77 Owing to the hyperglycaemic side effects the EMA has currently advised to limit the use of pasireotide LAR as a second-line agent for patients inadequately controlled with first-generation LA-SSAs.
Oral octreotide
Recently a new oral octreotide formulation (Octreolin®) has been developed which enables intestinal absorption of octreotide with limited intestinal bioavailability. The results of a phase III multicentre trial on the efficacy of oral octreotide showed that switching from injectable LA-SSA to the oral formulation can effectively maintain biochemical control in 65% of cases after 13 months.78
In conclusion, optimal care of acromegaly patients should be achieved by a tailored treatment approach that is based on pituitary tumour characteristics, GH and IGF-I levels and patient comorbidities plus the availability of a multidisciplinary team of experts in experienced centres. Serum IGF-I and GH levels should be measured by a validated assay in a dedicated endocrine laboratory, and clinicians should be aware of the various assay pitfalls. Currently, long-acting somatostatin analogues are the first line of medical treatment for acromegaly and surgery is the primary treatment option when the tumour is resectable, provided an experienced neurosurgeon is available. In case of a lack of response to a combination of LA-SSA, dopamine agonists and surgery, treatment with pegvisomant should be initiated. Pegvisomant is the most effective drug in achieving IGF-I normalisation to date, with or without co-treatment with LA-SSAs. The recently introduced second-generation multireceptor somatostatin analogue pasireotide seems to have a higher efficacy compared with the first-generation analogues octreotide and lanreotide, but a significantly higher incidence in hyperglycaemia has been reported during pasireotide treatment.
DISCLOSURES
The first author has nothing to declare. A.J. van der Lely is consultant for Novartis Pharma, Pfizer International and received grants from Novartis Pharma, Ipsen Pharma International and Pfizer International. S.J.C.M.M Neggers received research grants from Ipsen and Pfizer.
REFERENCES