DIAGNOSIS
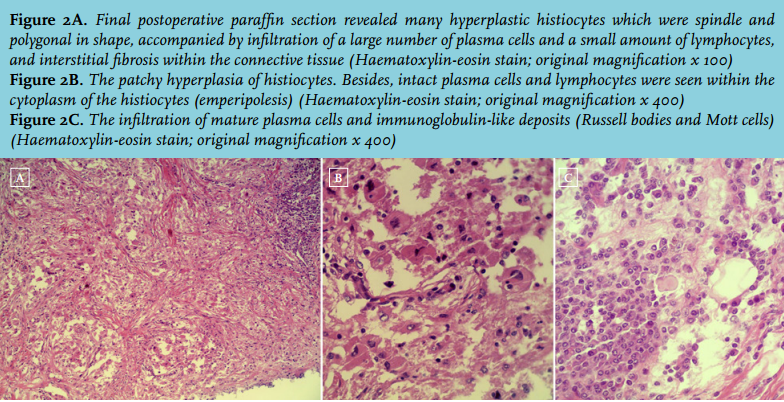
Final postoperative paraffin section revealed many hyperplastic histiocytes which were spindle and polygonal in shape, accompanied by infiltration of a large number of plasma cells and a small amount of lymphocytes, and interstitial fibrosis within the connective tissue (figure 2A). Intact plasma cells and lymphocytes were seen within the cytoplasm of the histiocytes (emperipolesis) (figure 2B). The infiltration of mature plasma cells and immunoglobulin-like deposits (Russell bodies and Mott cells) were also seen clearly under the microscope (figure 2C). A conclusive diagnosis of Rosai-Dorfman disease (RDD) of the left thigh was achieved. Immunohistochemistry staining showed that the lesions were positive for S-100, CD163, α1-antichymotrypsin, α1-antitrypsin and CD38, partly positive for CD68, λ and κ, but negative for CD1a. Immunohistochemistry staining also showed that the positive cell population of Ki-67 was < 2%. These findings also supported the diagnosis of RDD. The postoperative course was uneventful.
RDD, also known as sinus histiocytosis with massive lymphadenopathy (SHML), was first described by Rosai and Dorfman in 1969.1 RDD is a rare and non-neoplastic histiocytic proliferative disorder of unknown cause. It is considered to be a benign and self-limited disease, but it can also have an aggressive clinical course.2-4 RDD commonly occurs in adolescents or young adults. The typical clinical presentation of RDD is usually painless, bilateral cervical lymphadenopathy. Other clinical findings of RDD include fever, leukocytosis, anaemia, elevated ESR and hypergammaglobulinaemia.5,6 Extranodal involvements of RDD occur in 43% of all patients with lymphadenopathy.7 The extranodal sites include the skin, soft tissue, upper respiratory tract, orbit and bone. RDD located in the fat layer of the thigh as only an extranodal involvement without lymphadenopathy is an unusual presentation.
The definitive diagnosis of RDD relies on histopathological examination, and the immunohistochemical characteristics of the lesions contribute to determining the final diagnosis. Pathologically, RDD is characterised by a benign histiocytic proliferation with frequent lymphocytophagocytosis (emperipolesis).6,7 RDD must be differentiated pathologically from other lesions, such as lymphoma and plasmacytoma, fibro-inflammatory lesion, Langerhans cell histiocytosis and other histiocytosis. In some cases of RDD, obvious hyperplasia of lymphocytes or plasma cells resembles the appearance of lymphoma or plasmacytoma. However, the hyperplastic lymphocytes or plasma cells are of polyclonal origin, and simultaneously express λ and κ by immunohistochemistry. Furthermore, nuclear divisions are seldom seen in the RDD. While nuclear divisions are more commonly seen in the lymphoma or plasmacytoma, and their lymphocytes or plasma cells are of monoclonal origin. Thus, they can be ruled out. RDD may mimic the appearance of a fibro-inflammatory lesion. But this disease can be quickly ruled out because the histiocytes of a fibro-inflammatory lesion do not show emperipolesis and are negative for S-100. Langerhans cells are generally smaller than RDD cells. In Langerhans cell histiocytosis, the reniform nuclei with an obvious nuclear groove are usually observed (unlike the round nuclei of RDD cells), and emperipolesis is very rarely observed. Furthermore, Langerhans cells ultrastructurally contain characteristic Birbeck’s granules and are positive for CD1a (unlike RDD cells which are negative for CD1a). The treatment of RDD included surgical resection with or without chemotherapy, radiotherapy or corticosteroids. Our patient only underwent extended surgical resection of a mass in her left thigh without further treatment. She was followed up for four years and three months after surgery with a good prognosis.
REFERENCES