KEYWORDS
Haemophilia A, acquired, immunosuppressive agents, haemostasis

INTRODUCTION
Acquired haemophilia is a rare haemorrhagic disorder that results from the immune-mediated production of auto-antibodies to coagulation factors. The most common auto-antibodies that affect clotting factor activity and lead to a bleeding disorder are directed against and interfere with factor VIII (acquired haemophilia A). The reported incidence of this disease is approximately 1.5 per million.1 The most common conditions associated with the development of acquired factor VIII inhibitors are rheumatic disease, malignancy, some drugs (penicillin, sulphonamides, phenytoin), and the postpartum period; however, most cases are idiopathic (approximately 52%).2 Acquired haemophilia as a cause of post-procedural bleeding episodes is a potentially life-threatening disease in patients without a previous diagnosis of a bleeding disorder. Prompt diagnosis is crucial to initiate the appropriate treatment strategies based on the optimal haemostatic therapy and elimination of the inhibitor by immunosuppressive therapy. Here, we report a patient with delayed but profound postsurgical haemorrhage due to acquired haemophilia A, and illustrate that patience is necessary to achieve eradication of the auto-antibodies in order to achieve sustained control of the bleeding.
CASE REPORT
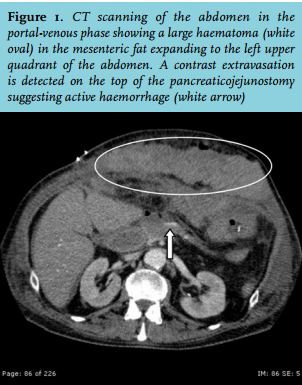
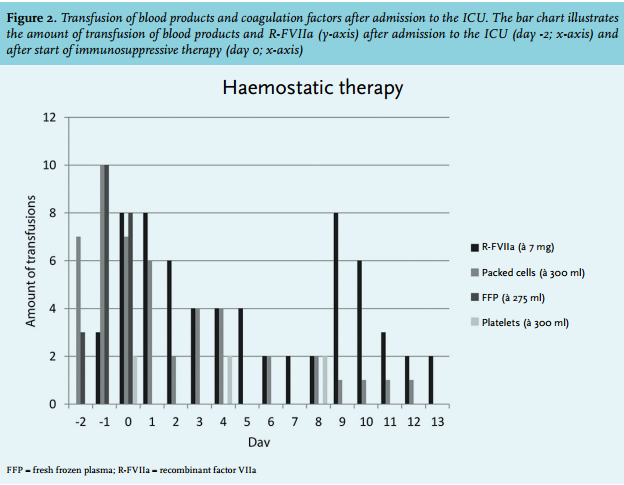
A 67-year-old man was diagnosed with pancreatic cancer for which he underwent a conventional pancreaticoduodenectomy (pT3N1M0, R0 resection), without receiving adjuvant systemic chemotherapy. The postoperative course was complicated by an enterocutaneous fistula, which was surgically resected two months after the first operation. Intraoperatively, there was an obvious oozing which was controlled by haemostatic gauzes. The early postoperative period was characterised by slowly decreasing haemoglobin levels. On the seventh postoperative day, the clinical condition of the patient abruptly deteriorated, characterised by circulatory shock. Computed tomography (CT) scan of the abdomen demonstrated a large mesenteric haematoma (figure 1), and active bleeding from one of the branches of the superior mesenteric artery, which was successfully embolised. Laboratory testing on the intensive care unit (ICU) revealed a haemoglobin level of 4.9 mmol/l, platelet count 375 x 109/l, a prothrombin time (PT) of 23 seconds, and an activated partial thromboplastin time (aPTT) of 99 seconds. Despite multiple transfusions (red blood cells, platelets, fresh frozen plasma, activated prothrombin complex concentrate, tranexamic acid, and vitamin K (figure 2)), the patient developed progressive circulatory shock. A second CT angiography showed a new blush in a distal branch of the right internal mammary artery which was also embolised. After that, an abdominal compartment syndrome developed, which required surgical decompression. Multiple transfusions of blood products and vitamin K normalised the PT; however, the aPTT remained prolonged.
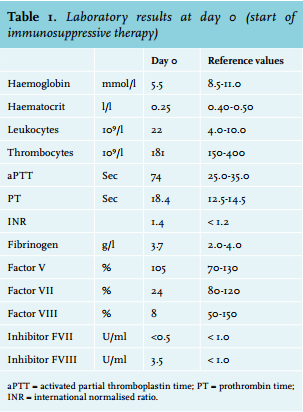
At this stage acquired haemophilia A was suspected and a mixing test was performed which confirmed the presence of an inhibitor. Further investigations showed a factor VIII activity of 8% (normal range 55-200%), and the Bethesda assay revealed a low titre of the present inhibitor against factor VIII (3.5 Bethesda units (BU)) (table 1). Factor VII activity (24%) was also reduced due to consumption caused by severe haemorrhage and a vitamin K deficiency. No inhibitor against factor VII was measured. Retrospectively, coagulation tests at the time of the pancreaticoduodenectomy showed normal clotting times. In consultation with the Van Creveld Clinic, the haemophilia treatment centre of the University Medical Center Utrecht, the patient was initially treated with factor VIII (AaFact® 50 IE/kg bolus, followed by 50 IE/kg/24 h). However, the suppletion of factor VIII was discontinued the next day due to a low yield in factor VIII activity (peak rise of 13%). Therefore, recombinant factor VIIa (R-FVIIa) (NovoSeven®) was started in a dose of 90 µg/kg (figure 2).
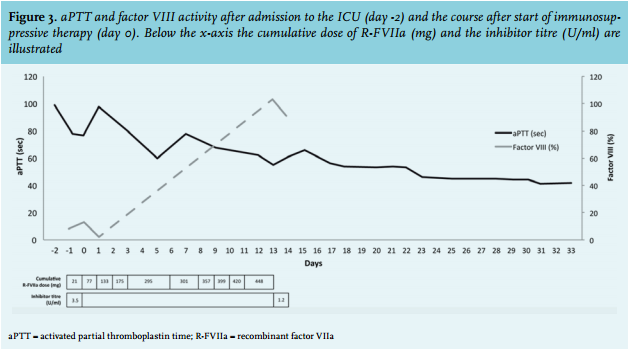
To promote elimination of the factor VIII inhibitor, glucocorticoids were started (methylprednisone 1 gram/ day for three days, prednisone 80 mg/day thereafter) plus cyclophosphamide (100 mg/day). After the start of immunosuppressive therapy, the patient also received prophylactic cotrimoxazole. Three days later (fourth laparotomy), no bleeding focus was detected and the gauzes were removed. An intra-abdominal drain nearby the recurrent leakage of the pancreaticojejunostomy was left behind. Five days after the start of immunosuppressive therapy, the patient became less dependent on red blood cell transfusions and the haemoglobin level remained stable. We continued R-FVIIa in tapering doses for three more days. At day 13, factor VIII had normalised, the antibody titre declined (1.2 U/ml), and approximately 30 days after the start of immunosuppressive therapy the aPTT normalised (figure 3). There were no recurrent episodes of haemorrhage and the prednisone dose was slowly tapered by 10 mg each week. Unfortunately, 35 days after admission to the ICU the patient developed a rapidly progressive abdominal septic shock due to ongoing leakage of the pancreaticojejunostomy causing multiple, non-resolvable, intra-abdominal abscesses. Despite administration of broad-spectrum antibiotics and supportive treatment, refractory septic shock developed and the patient died. The next of kin did not give permission for autopsy.
DISCUSSION
Here we report a patient with delayed bleeding due to acquired haemophilia A, which is a rare auto-immune disorder. In most cases the cause is idiopathic; however, it may occur in association with malignancies and some perioperative drugs (e.g. antibiotics). Although malignancy was highly suspected to be the causative factor in our patient, the aPTT was not prolonged at the time of the first surgical intervention, which makes this cause most unlikely. An isolated prolonged aPTT has a limited differential diagnosis, and after excluding the use of heparin, the primary initial diagnostic test is a mixing test (adding normal plasma in a ratio of 1:1). Persistent prolongation of the aPTT indicates the presence of an inhibitor and the Bethesda assay should quantify the antibody titre. The disease is frequently diagnosed with considerable delay despite abnormal coagulation tests (median time to diagnosis 0-8 days).1 This suggests that many patients with severe bleeding and acquired haemophilia remain undiagnosed for a worrying length of time.
Treatment of acquired haemophilia can be divided into management of haemostasis and elimination of the inhibitor with immunosuppressive therapy. Although some patients (approximately 30%)2,3 do not require haemostatic treatment, in the majority of the patients with acquired haemophilia bleeding is an emergency. To control haemostasis in these patients, two strategies can be used. First using bypassing agents (R-FVIIa, factor eight inhibitor bypassing activity (FEIBA)), and second by increasing factor VIII levels (factor VIII concentrate and desmopressin). Bypassing agents, which act by directly restoring the platelet surface factor X activation, which is deficient in the absence of factor VIIIa, are recommended as first-line therapy because of their rapid action and high level of effectiveness.3,4 In patients with low factor VIII inhibitor titres on the other hand, high doses of factor VIII can be considered. However, as in our case, factor VIII administration may be ineffective, and to manage the severe haemorrhage, repeated administration of R-FVIIa should be initiated rapidly. The usual dose of R-FVIIa for treatment of acute bleeding is 90 to 120 µg/kg at two- to three-hour intervals until haemostasis is achieved. FEIBA is also an effective bypassing agent and comparable results to R-FVIIa are achieved at a dose of 75 IE/kg every 8-12 hours.5 Further dosing and lengthening of R-FVIIa and FEIBA should be based upon the patient’s clinical circumstances and expert opinion. In the treatment of our patient, the haemophilia treatment centre, the Van Creveld Clinic, was consulted.
The guidelines recommend immunosuppression as soon as the diagnosis has been confirmed.4 First-line immunosuppression is usually composed of steroids alone or steroids plus cytotoxic agents, although rituximab is increasingly being used either alone or in combination with other agents.6-8 Steroids with cyclophosphamide resulted in more stable complete remission (70%), compared with steroids alone (48%) or rituximab-based regimens (59%).9 The median time to complete remission was approximately five weeks for steroids with or without cyclophosphamide. Rituximab-based regimens are required for approximately twice as long, which exposes patients to a longer period of increased risk of bleeding. A literature review of 71 patients treated with rituximab and a variety of immunosuppressive agents found a response rate of more than 90%, but this study was prone to positive publication bias.10 Therefore, the current guidelines suggest that rituximab should only be used as a second-line agent in combination with steroids.10,11 Other options for the treatment of acquired haemophilia A are administration of intravenous immune globulin or the use of extracorporeal plasmapheresis. However, given the excellent results obtained with prednisone and cyclophosphamide, these treatments are not suggested as first line.
In consultation with the haemophilia treatment centre of the University Medical Center Utrecht, the Van Creveld Clinic, we treated our patient with steroids (1 mg/kg) in combination with cyclophosphamide (100 mg), which is the most commonly used regimen in the Netherlands. With this regimen, a starting biochemical and clinical response is expected approximately two weeks after initiating immunosuppressive therapy. The duration of treatment should be based on individual patient characteristics and biochemical responses. The primary goal of treatment should be monitored; however, since inhibitor titres drop very slowly following successful treatment, it is advisable to check the inhibitor titre every two to four weeks once immunosuppressive therapy has been started. Accurate monitoring of patients with acquired haemophilia A is necessary to detect an early relapse. The relapse rate after a first complete remission has been estimated at approximately 20%; 70% of such relapsing patients achieve a second complete remission.1
CONCLUSION
This case describes a patient undergoing a surgical procedure complicated by severe intra-abdominal haemorrhage due to undiagnosed acquired haemophilia A. The finding of an isolated prolonged aPTT together with the presence of ongoing haemorrhage should trigger rapid diagnostic investigations for acquired haemophilia. The start of appropriate haemostatic treatment is crucial since delayed treatment may increase mortality. Similar to the observations in the registry data, our case emphasises that patience is necessary to achieve eradication of the auto-antibodies in order to control the risk of bleeding.
DISCLOSURES
The authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES