KEYWORDS
Cholesteryl ester storage disease, lysosomal acid lipase, familial hypercholesterolaemia, low-density lipoprotein cholesterol

INTRODUCTION
Cholesteryl ester storage disease (CESD), or lysosomal acid lipase deficiency (MIM #613497), is an autosomal recessive disease caused by mutations in the LIPA gene, encoding lysosomal acid lipase (LAL). LAL hydrolyses cholesteryl esters and triglycerides in lysosomes and a deficiency of this enzyme leads to intracellular accumulation of cholesteryl esters and triglycerides in hepatocytes, adrenal glands, intestine and cells of the macrophage-monocyte system.1 As a consequence, patients suffering from LAL deficiency are in general characterised by hepatomegaly, splenomegaly, diarrhoea and mixed hyperlipidaemia. Depending on the residual enzyme activity, LAL deficiency either results in the very severe and lethal Wolman disease (< 12 months of life) or the late-onset form, called CESD.1 CESD often remains undiagnosed until severe symptoms (i.e. liver failure and/or atherosclerotic disease) occur in adulthood.2 To date, ~140 CESD patients have been reported in literature while the prevalence of the disease has been estimated to vary between 1:40,000 and 1:400,000 individuals.2-4 The discrepancy in the reported and estimated number of patients might be caused by a large phenotypical variation of the disease and unawareness among medical professionals. We here present two family cases with substantially different clinical presentations of CESD, illustrating that this disease might be easily missed or remain undiagnosed.
CASE 1
A 23-year-old female was diagnosed with a clinical phenotype of primary hypercholesterolaemia during a routine cholesterol screening at work (total cholesterol 13.1 mmol/l; low-density lipoprotein-cholesterol (LDL-C 10.6 mmol/l; high-density lipoprotein-cholesterol (HDL-C) 1.75 mmol/l; triglycerides 1.69 mmol/l). She did not have any symptoms and physical examination was normal except for a systolic cardiac murmur (grade IV/ VI) due to a supravalvular aortic stenosis. Subsequent laboratory analysis only revealed mild increases in alanine aminotransferase (ALAT) levels in the proband (56 U/l; reference: 0-45 U/l).4 Family screening showed that both of her siblings were also affected while lipid levels were normal in their parents.4 However, no mutation was identified in one of the well-annotated genes for either autosomal dominant or recessive hypercholesterolaemia, (LDLR, APOB, PCSK9 and LDLRAP). In order to find a mutation in an unknown gene as cause of the hypercholesterolaemic phenotype, we sequenced all protein coding regions of the genome (exome sequencing). Unexpectedly, homozygosity for the exon 8 splice junction (E8SJ = c.G934A) mutation in LIPA was found.5 The patients were therefore diagnosed with CESD which was biochemically confirmed by a residual LAL activity of 8% in the proband.4 Both siblings of the proband were also found to be homozygous for the E8SJ mutation and all three patients were treated with statin therapy resulting in an impressive decrease in LDL-C levels by 73 to 88% in the proband and her siblings.4 We performed magnetic resonance spectroscopy in order to determine the consequences of the LIPA mutations on cholesteryl ester storage accumulation in the liver. Both the proband and her two siblings turned out to have substantial hepatic cholesteryl ester accumulation, but hepatosplenomegaly was not observed.4 This case was previously published by Stitziel and co-workers.4
CASE 2
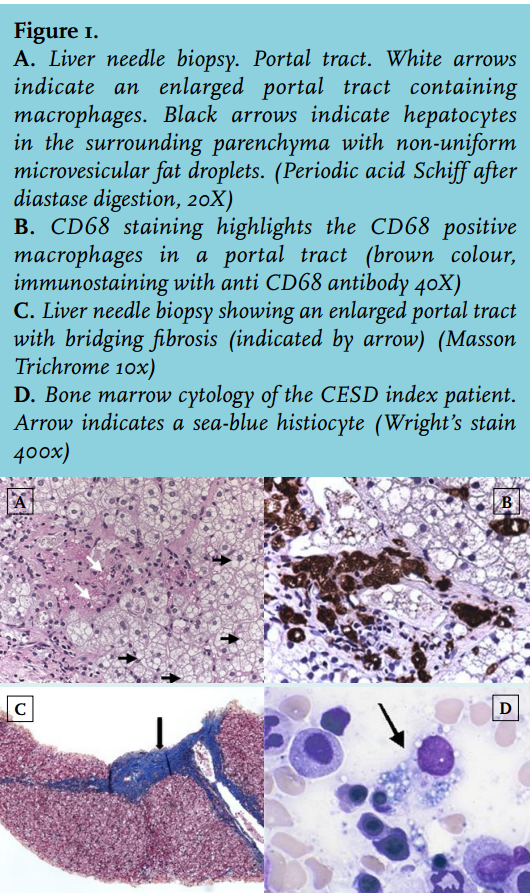
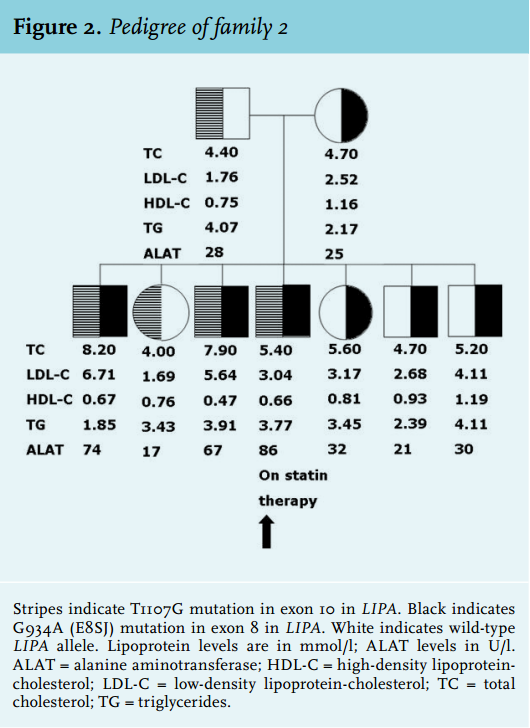
A 34-year-old male of Dutch/Indonesian descent was admitted to the hospital because of upper abdominal pain, diarrhoea and intestinal blood loss. His past medical history was relevant for hepatosplenomegaly of unknown origin since childhood, for which no follow-up had taken place. A surgical biopsy obtained when he was 3 years old remained inconclusive. Hepatosplenomegaly without signs of chronic liver failure was confirmed upon physical examination. Routine laboratory tests for primary liver disease were all normal except for increased ALAT levels (86 U/l while treated with a statin). Hypercholesterolaemia (total cholesterol 9.0 mmol/l, total cholesterol/HDL ratio 16.3) was also present. Stool weights were normal with normal fat content. Varices at the rectosigmoid junction were found upon colonoscopy, and gastroscopy performed at a later stage confirmed the diagnosis of portal hypertension (grade I). Liver biopsy (figure 1A-C) showed periportal and bridging fibrosis, without signs of cirrhosis. The hepatocytes were enlarged and contained non-uniform microvesiculae. Foamy macrophages were seen both in the portal tracts and in the parenchyma. Bone marrow cytology showed few, but clearly distinguishable, ‘sea-blue histiocytes’ and vacuolated macrophages, indicators of, among others, a lysosomal storage disease (figure 1D).5-7 In line, chitotriosidase levels were also increased (325 nmol/ml/h; reference < 200 nmol/ml/h), indicating macrophage accumulation.5,8 Microscopy findings were consistent with the diagnosis of Niemann-Pick disease (type B or C) but the finding of normal sphyngomyelinase activity in leucocytes and the negative filipin staining of cultured fibroblasts refuted these diagnoses. These results, combined with reduced cholesterol esterification, did not, however, exclude CESD. Therefore, LAL activity was subsequently measured in fibroblast cultures and was found to be strongly decreased (0.2 nmol/h per mg fibroblast protein in the proband versus 8-15 nmol/h per mg fibroblast protein in control cultures). Since mutations in LIPA are known to cause reduced LAL activity and, as a consequence CESD, we used targeted DNA diagnostics (RT-PCR) of the LIPA gene to confirm the diagnosis. This revealed compound heterozygosity for the E8SJ mutation and the T1107G mutation in exon 10 in LIPA. Two brothers were also diagnosed with CESD (figure 2). Neither of them had sought medical attention for CESD-related symptoms and might have remained undiagnosed if the proband had not been identified as a CESD patient. The portal hypertension in the proband was successfully treated with propanolol. The diarrhoea ceased after treatment with fibres (psyllium). After confirmation of the diagnosis of CESD, treatment was started with a statin. Serum cholesterol and total cholesterol/HDL ratios decreased twofold.
DISCUSSION
We here describe two family cases of CESD. Case 1 presented with a typical phenotype of familial hypercholesterolaemia. In case 2, the combination of bone marrow cytology (‘sea-blue histiocytes’ and vacuolated macrophages), liver histology (microvesicular steatosis, bridging fibrosis and lipid laden macrophages), and a combined hyperlipidaemia finally led to the diagnosis of CESD. These cases illustrate the variability of phenotypes of patients with CESD and emphasise that in patients with hypercholesterolaemia of unknown origin or unexplained hepatosplenomegaly, LIPA mutations could be present. Moreover, our two cases clearly show the differential diagnostic pathways that can ultimately lead to the identification of a uniform molecular pathological substrate of a disease. It is of note that heterozygosity for LIPA mutations is not associated with clinical signs and symptoms. The E8SJ mutation is the most frequently found mutation in CESD patients, leading to skipping of exon 8 and resulting in only a fraction of the normal hLAL enzyme levels.9,10 The T1107G mutation in exon 10 of LIPA causes a premature stop codon that consequently results in the synthesis of a truncated LAL protein. Together with the exon 8 mutation this results in a nearly complete abolishment of hLAL activity in the compound heterozygotes. To the best of our knowledge, this mutation has not been previously described.2
Since elevated LDL-C and triglyceride levels as well as decreased HDL-C levels are associated with development of cardiovascular disease (CVD), CESD patients are likely to be at increased CVD risk.11 The low number of patients known thus far, however, preclude us from drawing firm conclusions in this regard. Although no specific therapy for CESD is available to date, current treatment of CESD patients comprises treatment of dyslipidaemia with statins, given the established beneficial effect on both lipids and CVD risk.11 The effect of statin therapy on the extent of hepatic cholesteryl-ester accumulation is, however, unknown.10
Sebelipase alpha (SBC-102; Synageva BioPharma Corporation, Lexington, Massachusetts, USA) is a recombinant human LAL enzyme that is currently in the clinical stage of development and that has been tested in nine CESD patients to date.12,13 In this population, sebelipase alpha has shown to significantly reduce ALAT and aspartate aminotransferase (ASAT) levels by 52% (44 U/l) and 36% (20 U/l), respectively after 12 weeks (4 weekly infusions followed by 4 bi-weekly infusions of 0.35 mg/ kg – 3 mg/kg).13 These decreases were shown to sustain for up to 52 weeks of treatment in seven patients, where ALAT levels were decreased by 58% (49 U/l) and ASAT levels were decreased by 40% (23 U/l) (p = 0.016) from baseline (4 weekly infusions followed by 24 bi-weekly infusions). Besides the reductions in transaminase levels, sebelipase alpha has also shown to significantly reduce lipid and lipoprotein levels. After 52 weeks of treatment, total cholesterol levels decreased by 1.84 ± 0.80 mmol/l (p = 0.016), LDL-C decreased by 1.89 ± 0.80 mmol/l (p = 0.016) and triglyceride levels decreased by 0.81 ± 0.41 mmol/l (p = 0.047). Infusion reactions were only observed in one patient, who was finally successfully re-challenged.13 Sebelipase alpha is currently being tested in a placebocontrolled phase III trial (ARISE; NCT 01757184).
The non-specific phenotypic characteristics of CESD and the (partial) overlap of phenotypic features with other cardiovascular, liver and metabolic diseases illustrate the difficulty in diagnosing patients with CESD and suggest that patients with this disease might be missed or remain undiagnosed. This is supported by the large discrepancy between the number of reported CESD cases in the literature and the estimated prevalence of this disease.3,10 The large variability of CESD phenotypes, as illustrated in this case report, shows that more insight into the natural clinical course of homozygosity/compound heterozygosity for LIPA is warranted, since this will influence the identification of patients who will have benefit from future enzyme replacement therapy.
ACKNOWLEDGEMENTS
GKH is a recipient of a Veni grant (project number 91612122) from the Netherlands Organisation for Scientific Research.
DISCLOSURES
Dr. Hovingh received lecture fees from Pfizer, Sanofi, Regeneron, Synageva, Amgen, Roche, and Genzyme. Dr. Alleman received lecture fees from Orphan Europe. The other authors declare that they have no conflicts of interest.
REFERENCES