KEYWORDS
Hemolytic anemia, blood smear, next generation sequencing

INTRODUCTION
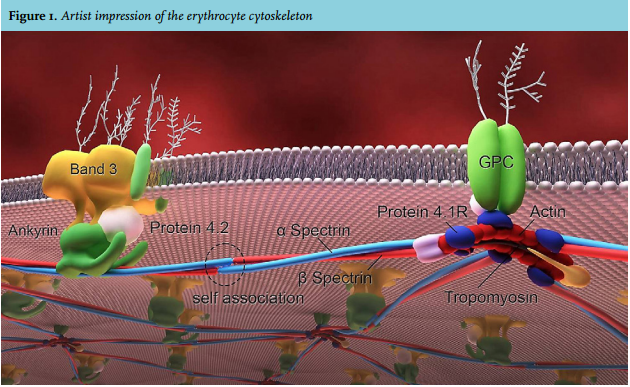
Many skills in jobs, art forms and sports are based on the application, repetition and training of elementary principles. The following case report reminds us to pursue the basic steps in elucidating the cause in a patient with haemolytic anaemia. Erythrocyte membrane abnormalities are a well-known cause of hereditary haemolytic anaemia. The specific make-up of the red cell membrane accounts for its remarkable deformability and enables the cell to pass through small capillaries with a diameter of only one-third of its own. The membrane consists of a lipid bilayer in which transmembrane proteins are located which anchor the bilayer to an intracellular cytoskeleton. The latter forms a two-dimensional hexagonal filamentous meshwork, mainly consisting of spectrin tetramers (figure 1). Spectrin tetramers consist of alpha- and beta-spectrin protein, linked in a head-to-head arrangement, and encoded by the SPTA1 and SPTB genes. Another prominent membrane protein is band 3, encoded by the SLC4A1 gene. Band 3 is located in the erythrocyte plasma membrane and is part of a multiprotein complex that acts as an attachment site for the cytoskeleton. Abnormalities of each component of the cytoskeleton and anchoring proteins compromise their interactions, often leading to characteristic shape changes.1
CASE PRESENTATION
A 20-year-old Caucasian woman presented with jaundice. Her previous history included an episode of haemolytic anaemia three years earlier during a parvo B19 infection. At that time laboratory investigation revealed no haemoglobinopathy and no deficiencies of glucose-6-phosphate dehydrogenase or pyruvate kinase. Erythrocyte membrane protein analysis revealed ‘normal’ signals for spectrin (RIA) and Band 3 (EMA/FACS). One year later a second haemolytic episode occurred during an Epstein-Barr virus infection. Five days before consultation she became acutely ill with nausea, vomiting and diarrhoea when travelling in Spain. Symptoms abated spontaneously, but she became jaundiced. On examination she was not acutely ill, but icteric. Results of laboratory investigations pointed to haemolysis as the cause of the jaundice (table 1).
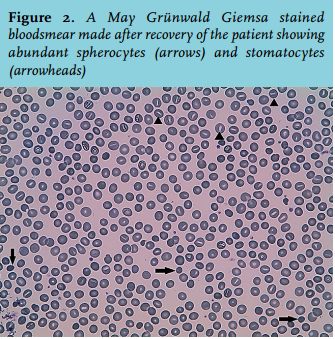
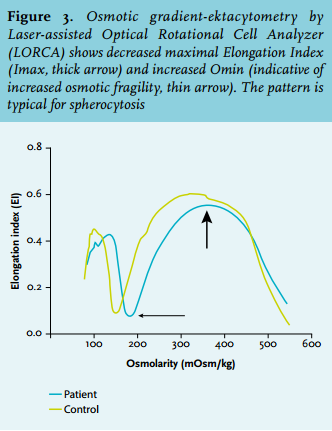
A presumptive diagnosis of mild infectious enterocolitis with episodic nonimmune haemolysis was made. A new systematic review was performed. The persistently decreased level of haptoglobin, increased bilirubin, and reticulocytosis were indicative of mild chronic haemolysis. The blood smear disclosed abnormal erythrocyte morphology with prominent presence of spherocytes and stomatocytes (figure 2). Osmotic gradient ektacytometry showed decreased EImax and increased Omin, indicative for spherocytosis (figure 3).
Based on these results a novel next-generation sequencing (NGS) technique was applied to detect mutations in the seven genes most commonly associated with erythrocyte membrane disorders (SPTA1, SPTB, SLC4A1, ANK1, EPB41, EPB42, and RHAG). This analysis revealed compound heterozygosity for two pathogenic SPTA1 gene mutations: c.2806-13T>G (α-spectrinSt Claude) and c.4339-99C>T (α-spectrinLEPRA). These mutations have previously been associated with autosomal recessively inherited poikilocytic anaemia and spherocytosis, respectively. In addition, the patient was found to be heterozygous for a c.118G>A, p.(Glu40Lys) missense mutation in SLC4A1 which is associated with autosomal recessively inherited spherocytosis. The two α-spectrin mutations were present on separate alleles (in trans) as confirmed by DNA analysis of the parents.
DISCUSSION
Examination of the peripheral blood smear was the key in establishing the correct diagnosis in this patient, despite normal results for red cell membrane disorder markers in previous investigations. In addition, osmotic gradient ektacytometry was performed which is the ‘gold’ standard for the detection of various disorders of the red cell membrane. By this method erythrocytes are exposed to both shear stress (by centrifugal force) and gradually changing osmotic conditions. These induced changes in the shape of the red cells are detected and render characteristic deformability profiles for elliptocytosis, spherocytosis, and stomatocytosis.2 Heterogeneity in the clinical severity and erythrocyte morphology of the hereditary haemolytic anaemias due to defective membranes is the rule as different proteins are involved in conjunction with a variety of genetic causes: deletions, nonsense, missense, null and low expression mutations and splicing variants. The SLC4A1 c.118G>A mutation was first reported by Rybicki et al. and the protein harbouring the
consequent p.(Glu40Lys) substitution was named band 3Montefiore. 3 Their propositus was homozygous for this mutation and had hereditary spherocytosis. The defective band 3 protein probably resulted in faulty interactions with protein 4.2 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Relatives who were heterozygous for the mutation were clinically unaffected. Whether the heterozygous state for this mutation is clinically relevant for the current case is uncertain, but an additive effect on the presentation of clinical features cannot be excluded.
Most mutations in the SPTA1 gene affect the self-association site of the α- and β-spectrin molecules, leading to elliptocytosis. However, mutation at other sites can lead to pyropoikilocytosis and spherocytosis. The SPTA1St Claude allele was first reported by Fournier et al. 4 Their homozygous propositus was symptomatic whereas the heterozygous parents were clinically and biochemically unaffected. The SPTA1LEPRA allele was reported by Wichterle et al. 5 Their propositus was compound heterozygous for SPTA1LEPRA and SPTA1PRAGUE causing severe spherocytic haemolytic anaemia. The parents, heterozygous carriers of either of these mutations, were asymptomatic. Intriguingly, apart from spherocytosis our patient showed a large number of stomatocytes on the blood smear. This unique appearance may be related to co-inheritance of the band 3Montefiore allele.
 It is highly likely that the mutations in spectrin and band 3 are responsible for the spherocytosis / stomatocytosis and the corresponding subclinical haemolysis. In patients with such membrane alterations intercurrent infections can exacerbate the haemolysis and provoke haemolytic crises. At the age of 15 and 16 years she had clinical jaundice which was attributed to haemolytic anaemia as part of serologically proven parvo B19 and Epstein-Barr virus infections, respectively, as the clinicians were unaware of the presence of structural changes in the red cell membrane. The last icteric episode occurred during an acute, self-limiting infectious enterocolitis (traveller’s diarrhoea). It is, however, remarkable that she had no history of haemolytic crisis earlier in life since she must have suffered from other infectious childhood diseases. Molecular analysis of the genes encoding red blood cell membrane proteins has long been hampered due to the size and number of the genes involved. With the advent of NGS techniques it has now become feasible to analyse many genes in parallel with high quality. For erythrocyte membrane disorders, the DNA diagnostics section of the UMC Utrecht has implemented NGS-based sequencing of seven genes known to be causally involved: SPTA1, SPTB, ANK1, SLC4A1, EPB41, EPB42 and RHAG. This gene panel analysis was validated and accredited under the ISO15189 standard and yields a mutation detection sensitivity of > 95% in the seven genes analysed. As such, a leap forward has been made in the facilitation of molecular genetic diagnostic analysis of patients with suspected erythrocyte membrane disorders. The fact that spectrin and expression of Band 3 protein on the erythrocyte membrane surface were previously determined as normal shows that a normal amount does not imply a normal structure and function of the protein.
It is highly likely that the mutations in spectrin and band 3 are responsible for the spherocytosis / stomatocytosis and the corresponding subclinical haemolysis. In patients with such membrane alterations intercurrent infections can exacerbate the haemolysis and provoke haemolytic crises. At the age of 15 and 16 years she had clinical jaundice which was attributed to haemolytic anaemia as part of serologically proven parvo B19 and Epstein-Barr virus infections, respectively, as the clinicians were unaware of the presence of structural changes in the red cell membrane. The last icteric episode occurred during an acute, self-limiting infectious enterocolitis (traveller’s diarrhoea). It is, however, remarkable that she had no history of haemolytic crisis earlier in life since she must have suffered from other infectious childhood diseases. Molecular analysis of the genes encoding red blood cell membrane proteins has long been hampered due to the size and number of the genes involved. With the advent of NGS techniques it has now become feasible to analyse many genes in parallel with high quality. For erythrocyte membrane disorders, the DNA diagnostics section of the UMC Utrecht has implemented NGS-based sequencing of seven genes known to be causally involved: SPTA1, SPTB, ANK1, SLC4A1, EPB41, EPB42 and RHAG. This gene panel analysis was validated and accredited under the ISO15189 standard and yields a mutation detection sensitivity of > 95% in the seven genes analysed. As such, a leap forward has been made in the facilitation of molecular genetic diagnostic analysis of patients with suspected erythrocyte membrane disorders. The fact that spectrin and expression of Band 3 protein on the erythrocyte membrane surface were previously determined as normal shows that a normal amount does not imply a normal structure and function of the protein.
CONCLUSION
This case presentation illustrates that a ‘simple’ examination of the blood smear, a basic laboratory test, is still a good starting point from which more sophisticated tests can be requested. The fact that the ‘automated’ haemocytometry report mentions results of a (leukocyte) blood cell ‘differentiation’ does not imply that the morphology of the red cells has been investigated. Moreover, most current automated cell counters do not recognise red cell shape alterations. Even if there has already been an extensive academic workup this should not discourage other physicians from performing a new systematic analysis in a cold case. Interaction with clinical chemists and the progress in DNA technology comes to aid the clinicians and patients at the bedside.
DISCLOSURES
The authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES