KEYWORDS
Acquired haemophilia, HIV
INTRODUCTION
Acquired haemophilia A (AHA) is a rare coagulation disorder with a reported incidence of 1.20-1.48 million/ year.1,2 The condition is characterised by the production of autoantibodies to factor VIII (FVIII), leading to inhibition of FVIII. Early detection is essential, as most patients develop severe haemorrhage leading to considerable morbidity and mortality.3 The most commonly associated conditions are other autoimmune diseases, malignancies, pregnancy, specific pathogens (e.g., hepatitis B and C, human herpes virus-8), and drug exposure (including antibiotics, clopidogrel, and interferon), although in approximately 50% of patients, the appearance of these autoantibodies is idiopathic.3,4
Acquired FVIII inhibitors associated with human immunodeficiency virus (HIV) infection, either with or without hepatitis C virus (HCV) co-infection, have been described by several case reports in the literature.5-10 South Africa has the largest HIV epidemic in the world, with a prevalence of 18.9%.11 The rare association of AHA with HIV infection is therefore relatively more prevalent in South African patients and experience in how to treat them, essential. We present here a case series of the two HIV-positive patients, as well as the two HIV-negative patients, with AHA diagnosed and treated by the haematology unit at Groote Schuur Hospital in Cape Town, South Africa, from 2011-2019.
CASES
We suspected a diagnosis of AHA in all patients when a prolonged activated partial thromboplastin time (aPTT), which did not correct with mixing studies, was observed. Subsequently, we found decreased FVIII activity and presence of FVIII inhibitors, confirming the diagnosis. Platelet count and prothrombin time (PT) were characteristically normal in all patients. There is no formalised treatment protocol for AHA in South Africa and management is determined by local expertise, dependent on both availability and cost of drugs. Once haemostasis is achieved, the focus of therapy is inhibitor eradication. In our setting we endeavour to achieve immunosuppression with corticosteroids and an alkylator agent such as cyclophosphamide. As the public health sector in South Africa does have significant resource constraints, and administration of biologic immune modulators (e.g., rituximab), as well as bypassing agents, are generally reserved for refractory cases, due to their high cost. See table 1 for an overview of all patients.
HIV-positive patients
Patient 1
Patient 1 was a 43-year-old female, diagnosed with HIV and started on antiretroviral drugs (ARVs; stavudine, lamivudine, and efavirenz) two years before presentation. She was also receiving rifampicin, ethionamide, ethambutol, and pyrazinamide for isoniazid-resistant pulmonary tuberculosis, diagnosed two months prior to presenting. At presentation, her CD4 count was 118 cells/μl and HIV viral load (VL) was undetectable. She presented with bilateral knee haemarthrosis and easy bruising with a duration of two weeks. Initial laboratory investigations showed aPTT of 140.1 seconds (s), which did not correct on mixing studies, and haemoglobin (Hb) of 3 g/dl. Subsequently, we observed a FVIII inhibitor titre of 218 BU/ml and FVIII levels of 0 IU/dl. Testing for antinuclear antibody (ANA), anti-double stranded DNA antibodies (anti-dsDNA), lupus anticoagulant, and hepatitis B and C were all negative.
She received red cell transfusions and was given methylprednisolone intravenously (IV) at 500 mg/day for four days, which was then changed to oral prednisone at 1.5 mg/kg. The patient experienced no further bleeds, but a month later was given cyclophosphamide (1g IV weekly x 2 doses) as her aPTT, although much improved, was still prolonged and she still had a high FVIII inhibitor titre (47.7s and 54.56 BU/ml, respectively). One week later, her aPTT had increased again to 60 s; thus, chlorambucil at a dose of 6 mg daily was commenced.
Over the following months, corticosteroids were tapered and aPTT and FVIII inhibitor levels followed a downward trend. Nine months after commencing immunosuppressive therapy, inhibitors were absent and corticosteroids were stopped. All signs of bleeding, including the haemarthrosis, had fully resolved. Chlorambucil was continued for a year in total. The patient is seen for follow-up annually and has had no relapse to date – eight years after achieving remission.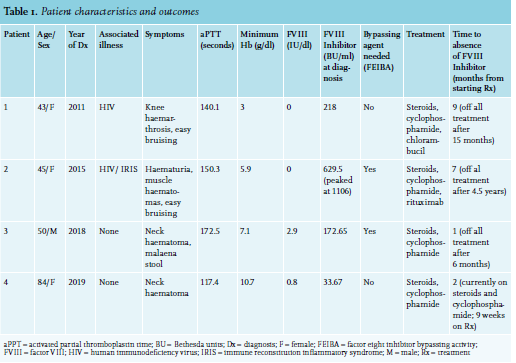
Patient 2
Patient 2 was a 45-year-old female who had been diagnosed with HIV and started on ARVs (tenofovir. emtricitabine, and efavirenz) two weeks prior to presenting. Her CD4 count at presentation was 144 cells/μl and HIV VL was undetectable at presentation. She presented with a several-day history of painless haematuria, easy bruising, and large haematomas to the lower limbs. Initial laboratory investigations showed aPTT of 150.3 s, which did not correct on mixing studies, and Hb of 5.9 g/dl. Subsequently, we observed a FVIII inhibitor titre of 629.5 BU/ml and FVIII levels of 0 IU/dl. ANA, anti-dsDNA, lupus anticoagulant and viral hepatitis studies were negative. We also identified tenofovir-induced nephropathy, which resolved when ARVs were changed to stavudine, lamivudine, and efavirenz.
Patient 2 received blood transfusions and one dose of methylprednisolone 125 mg IV, followed by prednisone at 1.5 mg/kg. Symptoms began resolving and aPTT improved until one week later, when her steroid dose was weaned to 1 mg/kg and she experienced a re-bleed and prolongation of aPPT to above 150 s. This resolved when the original steroid dose was resumed. Patient 2 experienced a re-bleed into her thigh muscle requiring a blood transfusion six weeks later, and cyclophosphamide 100 mg daily was added to her treatment. At this point, the FVIII inhibitor level had increased to 1103.6 BU/ml. She also required two doses (50 U/kg each) of FEIBA (Factor Eight Inhibitor Bypassing Activity), due to failure of the haematoma to resolve.
One month later, another attempt to wean her steroid and cyclophosphamide doses induced bilateral buttock haematomas, requiring multiple blood transfusions and, over the course of four days, five doses of FEIBA (50 U/kg each). At this stage, due to failure to wean immunosuppressive therapy as well as the need for repeated costly blood transfusions and use of bypassing agent, rituximab was given (375 mg/m2 per week for four doses). Several days after receiving the first dose of rituximab, the buttock haematoma worsened and she received eight more doses of FEIBA at 25-50 U/kg over the course of twelve days.
The patient was discharged and an attempt was made to taper the steroid dose again. The patient suffered one more re-bleed a month later, a thigh haematoma requiring one dose of FEIBA (50 U/kg). However, aPTT continued to improve and eventually, seven months after diagnosis and two months after rituximab administration, FVIII inhibitors were absent with aPTT and FVIII levels fully normalised. Immunosuppressive therapy dosage was tapered and, four-and-a-half years after diagnosis, discontinued. FVIII inhibitors did recur asymptomatically at low titres (1.48 and 0.49 BU/ml) on two occasions: six months after receiving rituximab at 1.48 BU/ml (at the time, the patient was on prednisone 0.5 mg/kg and the inhibitors resolved when the dose was increased to 0.6 mg/kg for three weeks); and again 11 months after rituximab (the patient was on prednisone 0.5 mg/kg; this time the inhibitors resolved without any dose adjustment two weeks later).
HIV-negative patients
Patient 3
Patient 3 was a 50-year-old male taking enalapril, hydrochlorothiazide, and amlodipine for hypertension for more than 10 years.
He presented with a several-day history of a midline neck haematoma and melena stool. aPTT was found to be 172.5 s and Hb 7.1 g/dl. FVIII inhibitors were present at 172.5 BU/ml and FVIII levels were 2.9 IU/dl. ANA, anti-dsDNA antibodies, anti-cardiolipin antibodies, HIV, and hepatitis B and C testing were all negative. Lupus anticoagulant was weakly positive: patient/control ratio 1.28 (normal < 1.2). The patient received FEIBA 50 U/kg daily for three days and the neck haematoma stabilised (the airway was never compromised) and melena resolved. Tranexamic acid was also given (1 g IV three times daily). He was started on prednisone 1.5 mg/kg and cyclophosphamide 100 mg daily. His aPTT decreased to 39 s the following week and he was discharged on the above medication. An absent inhibitor screen was found a month after initiating treatment and his immunosuppressive medication was tapered over the following six months with no recurrence of symptoms.
Patient 4
Patient 4 was an 84-year-old female with a long history of hypertension, dyslipidaemia, chronic obstructive pulmonary disease (COPD), and hypothyroidism of unknown aetiology. Her chronic medication was amlodipine, furosemide, and simvastatin; inhaled salbutamol, budesonide, and levothyroxine. She presented with a six-week history of a sub-lingual haematoma that had spread to the anterior neck, but was fortunately not compromising the airway. aPTT was 117.4 s and Hb 10.7 g/dl. FVIII inhibitors were present at 33.67 BU/ml and FVIII levels were 0.8 IU/dl. No other testing was done for autoimmunity. She was started on methylprednisolone 80 mg IV daily and the haematoma soon began improving. This was changed to prednisone 1 mg/kg and cyclophosphamide 100 mg daily, one week later. The haematoma resolved with no recurrence of symptoms and FVIII inhibitors were absent two months after initiating therapy. The patient is still on immunosuppression therapy nine weeks after being discharged, with dosage being tapered.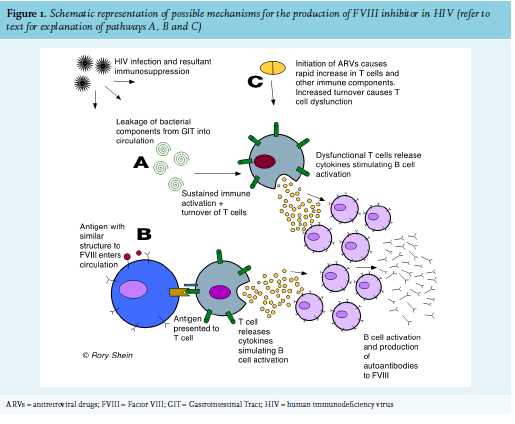 DISCUSSION
DISCUSSION
The precise mechanism for the appearance of FVIII inhibitors, usually a population of polyclonal IgG autoantibodies directed to FVIII12, is not known. In HIV, the breakdown of T-cell immune regulation and subsequent immune dysfunction allows for the development of autoimmunity, and numerous autoimmune conditions are associated with the disease. Various mechanisms of how this occurs have been postulated, including (figure 1):
 A. Long-term, sustained immune activation (due to leakage of bacterial components through the gut mucosa, caused by a dramatic decline in CD4+ T lymphocytes at this site seen early in HIV infection13), causing increased turnover and dysfunction of non-infected T cells. This leads to abnormal production of cytokines, causing the activation of B cells and increased production of immunoglobulins.14
A. Long-term, sustained immune activation (due to leakage of bacterial components through the gut mucosa, caused by a dramatic decline in CD4+ T lymphocytes at this site seen early in HIV infection13), causing increased turnover and dysfunction of non-infected T cells. This leads to abnormal production of cytokines, causing the activation of B cells and increased production of immunoglobulins.14
B. Molecular mimicry of a self-antigen from an infectious agent, where T or B cells that react to pathogen-derived peptides cross react with host self-peptides.9
C. The immune dysfunction seen in patients with immune reconstitution inflammatory syndrome (IRIS) recently started on ARVs. This has been well described as a cause for autoimmune thyroid disease.15,16
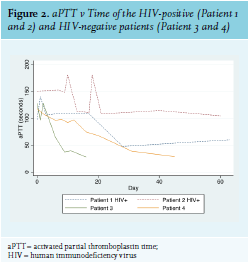
The development of AHA in Patient 2 could have been due to IRIS, as she had started ARVs two weeks prior to developing FVIII inhibitors and had experienced a rapid decline in viral load during that time. Most patients with AHA experience soft tissue, muscle, and mucosal (gastrointestinal tract, epistaxis, urinary tract) bleeds, while bleeding into the joints is rare (as opposed to the bleeding pattern in congenital haemophilia).3 Interestingly, this was not the case with Patient 1, whose primary sites of bleeding were both knee joints. The HIV-positive patients in this series had a significantly higher FVIII inhibitor titre and lower FVIII level than the HIV-negative patients, and had a much longer and more complicated course of treatment to induce their remission (see figure 2). This reflects existing data that shows that higher inhibitor titre and lower FVIII at diagnosis are associated with a longer time to remission.17 This correlates with other case reports of HIV-infected AHA patients, as well as with British and European multicentre registry-based studies of patients with AHA from all causes (see table 2). Inhibitors also developed at a younger age in the HIV-positive patients. All four patients in this case series survived and responded to immunosuppressive treatment.
As mentioned above, treatment of AHA patients in the SA public health sector is especially challenging due to resource constraints. Biologic immune modulators need special motivation and FEIBA is reserved for patients with non-resolving bleeds. As can be seen from Patient 2 above, the delay in administering rituximab led to slower resolution of symptoms and the need to use more FEIBA, ironically resulting in far more treatment costs than if it had been given sooner. This case series will serve to better inform the ‘balancing-act’ necessary to ensure efficient resource allocation while also optimising individual patient care. 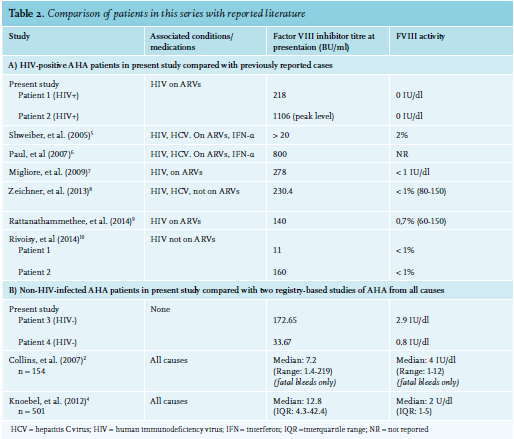 CONCLUSION
CONCLUSION
AHA is a rare autoimmune condition, and in our setting, associated with HIV infection in two of four pts. Early detection, as well as prompt initiation of treatment is essential, and for one patient in our series, timid initial use of scarce resources may have led to higher eventual costs. All patients survived without permanent morbidity. In this small cohort, the association with HIV infection seemed to portend a longer pathway to remission. This observation also correlates with other cases of HIV-associated AHA reported in the literature. Due to the rarity of the disease, it is vital to record and register cases to improve understanding and optimise future patient care.
ACKNOWLEDGEMENT
We would like to express thanks to Sister Anne-Louise Cruikshank, haemophilia nurse co-ordinator of the Western Cape Province, South Africa, for identifying these patients and providing on-going management.
DISCLOSURE
All authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES