KEYWORDS
Angioedema, cryoglobulinemia, MGRS, MGUS, monoclonal gammopathy
INTRODUCTION
Monoclonal gammopathy of undetermined significance (MGUS) is considered an asymptomatic precursor of malignant lymphoid disorders. In MGUS, an abnormal clone of a single plasma cell (precursor) produces a monoclonal immunoglobulin (M-protein) and/ or immunoglobulin light chains. The four diagnostic criteria for MGUS according to the International Myeloma Working Group are (1) M-protein lower than 30 g/l, (2) bone marrow plasma cells < 10% and low level of plasma cell infiltration in a trephine biopsy (if done), (3) no evidence of other B-cell proliferative disorders, and (4) no related organ or tissue impairment (CRAB-criteria: hypercalcaemia, renal insufficiency, anaemia, bone lesions).1
The prevalence of MGUS is 3.2% in people over the age of 50 years and 6.6% in people over the age of 80 years.2 The finding of MGUS is often unrelated to the patient’s primary medical problem. In general, patients with MGUS are not treated. However, despite the non-malignant nature of an ‘MGUS-like’ monoclonal gammopathy, it can cause significant morbidity. The next three cases will illustrate that MGUS can have severe consequences. 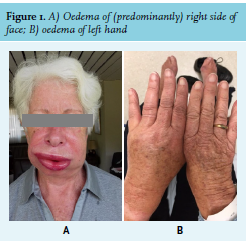
Case 1: Acquired C1-esterase inhibitor deficiency
A 78-year-old woman without relevant medical history was referred to the emergency department because of progressive unilateral oedema of the face, lips, and uvula since several hours (figure 1A). In the preceding days, she had had episodes of swelling of extremities (figure 1B). There was no fever, rash, or pruritus and the patient did not use medication. The patient was admitted to the Intensive Care Unit for observation. The swelling gradually disappeared after several hours. In the following three months, the patient presented almost every week to the emergency department because of similar episodes with unilateral swelling of the face, lips, and/ or extremities. Laboratory investigations (table 1) revealed low complement C4 and C1-esterase inhibitor deficiency. Antibodies against C1-esterase inhibitor were not detected. In addition, there was a presence of M-protein IgG-lambda and monoclonal free light chain lambda. A positron emission tomography-computed tomography (PET/CT) scan showed no lytic ossal lesions, and bone marrow biopsy immunophenotyping showed 5% monoclonal plasma cells. We diagnosed the patient with angioedema due to acquired C1-0esterase inhibitor deficiency secondary to a monoclonal gammopathy.
Attacks of angioedema were treated with C1-esterase inhibitor when there was risk of a compromised airway. Treatment with tranexamic acid 1000 mg had no effect on frequency and severity of angioedema episodes. Instead, treatment with danazol 200 mg, two times a day was started, after which, the patient had no more episodes of angioedema in the next 11 months. The dosage of danazol was gradually decreased to 100 mg once daily.
Seven months after the start of danazol, M-protein concentration had increased to 33 g/l and renewed bone marrow biopsy showed 23% plasma cells. Moreover, Bence Jone protein was detected and urinary sediment showed erythrocyte cylinders (table 1). The patient was diagnosed with high-risk smouldering myeloma (Revised International Staging System, R-ISS stage II). Treatment with lenalidomide and dexamethasone (cycles of 28 days with lenalidomide from days 1 to 21 and dexamethasone on days 1, 8, 15, and 22) and pamidronic acid (once per four weeks) was started. Eight weeks after start of treatment (i.e., two cycles), M-protein concentration had decreased from 33 to 8 g/l, and danazol was ceased. Another eight weeks later (after four cycles), M-protein concentration was < 2 g/l and C1-esterase inhibitor levels had normalised. Until present day (six months after start of treatment), the patient did not have an attack of angioedema and serum M-protein remains undetectable. 
Case 2: Cryoglobulinemia
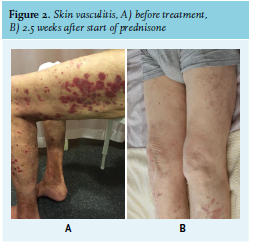
An 83-year-old man with a history of myocardial infarction (1989), peripheral vascular disease and femoral-popliteal bypass (2018) was referred to the nephrology outpatient department with progressive renal insufficiency and nephrotic-range proteinuria (8.20 g/24hr). Within seven months, the estimated glomerular filtration rate (eGFR) had gradually decreased from 64 to 16 ml/min/1.73 m2. Furthermore, the patient had skin lesions that had started as episodic purpura on the legs seven months earlier, but had progressed to permanent non-pruritic palpable conflating purpura on the trunk and all extremities (figure 2A). Laboratory investigations (table 1) revealed monoclonal free light chain type kappa, presence of cryoglobulins (monoclonal IgM kappa and polyclonal IgG and IgA), and low complement C3 and C4. Unfortunately, rheumatoid factor was not determined. Hepatitis B and C serology were negative. There were no symptoms or findings suggesting systemic disease such as Sjögren syndrome. A kidney biopsy showed glomerulonephritis with deposition of IgM, kappa, and C3, and a skin biopsy showed leucocytoclastic vasculitis with deposition of C3 in the vascular wall. Bone marrow examination revealed 1% monoclonal B cells, without increased percentage of plasma cells or lymphocytes. We diagnosed the patient with cryoglobulinemia type II with skin vasculitis and glomerulonephritis secondary to a monoclonal gammopathy. We started treatment with prednisone 60 mg per day. This had a spectacular response. After only 2.5 weeks, the skin lesions had almost vanished (figure 2B) and the eGFR had improved from 16 to 45 ml/min/1.73 m2. Furthermore, cryoglobulins were no longer detectable and the kappa/ lambda light chain ratio had normalised. Because of this quick and complete response, and frailty of the patient, we decided to withhold targeted treatment of the monoclonal B cells with rituximab.
In the following seven months, the prednisone was slowly tapered to a total daily dose of 15 mg. At that point, kappa/ lambda light chain ratio increased to 2.0 (kappa free light chain 45 mg/l), cryoglobulins were detectable again, and proteinuria increased somewhat (urine albumin/creatinine ratio 102 mg/mmol). Despite these signs of relapse, we decided to continue monotherapy with prednisone 15 mg, because of stable kidney function, absence of skin lesions, and increased frailty in the meantime (amputation of the upper leg due to acute ischemia and de novo chronic obstructive pulmonary disease). The patient died several weeks later due to this comorbidity.
Case 3: Monoclonal gammopathy of renal significance
An 86-year-old man with a history of hypertension and stable chronic kidney disease was referred to the nephrologist because of progressive decrease in kidney function (increase of serum creatinine from 115 to 144 μmol/l in one year). Physical examination was normal except for a blood pressure of 160/80 mmHg despite use of losartan 100 mg daily. Laboratory results (table 1) showed progressive kidney failure with nephrotic-range proteinuria and presence of monoclonal IgG kappa and free light chain kappa. Bone marrow biopsy showed 6% plasma cells. PET-CT revealed no osteolytic lesions. A renal biopsy showed mesangiocapillary glomerulopathy with signs of thrombotic microangiopathy and tubulopathy secondary to paraproteinemia, and 40% sclerosed glomeruli and intima fibrosis of arterial branches. We diagnosed the patient with glomerulonephritis with nephrotic syndrome caused by monoclonal gammopathy of renal significance (MGRS). Because of high age, lack of symptomatology, and relatively mild expression, we decided on a wait-and-see policy.
Two years later, the patient complained of back pain. A CT scan revealed multiple osteolytic lesions in skull and spine A renewed bone marrow biopsy again showed 5-10% plasma cells (the same as two years earlier). Serum creatinine had increased to 244 μmol/l (Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) for eGFR 20 ml/ min/1.73 m2 ). The quantity of M-protein, kappa/lambda free light chain ratio, and urine albumin/creatinine ratio were lower than two years earlier (12 g/l, 21, and 262 mg/mmol, respectively). Although plasma cells in the bone marrow were only 5-10%, patient was treated as having symptomatic multiple myeloma. Treatment with dexamethasone 20 mg per week, lenalidomide 5 mg per day, and pamidronic acid 30 mg once per month was started. After three months, M-protein had decreased to 8 g/l and kappa/lambda free light chain ratio to 7.5. However, serum creatinine and proteinuria had not improved and patient had developed a pancytopenia. As improvement of renal function was not expected with continuing therapy, and whereas side effects as pancytopenia would worsen, it was decided to cease treatment. In the six months thereafter, clinical condition and renal function gradually decreased (serum creatinine 339 μmol/l/1.73 m2 , CKD-EPI eGFR 13 ml/min/1.73 m2 ), while kappa/lambda free light chain ratio rose to 19. Because of immobility and poor prognosis, the patient wished to transfer care to his general practitioner for palliative care.
DISCUSSION
We describe three patients with a monoclonal gammopathy that can be classified as monoclonal gammopathy of undetermined significance (MGUS). However, the diagnosis MGUS is not applicable, as the significance of the monoclonal gammopathy in these patients is not ‘undetermined’, but pathologic.
Acquired C1-esterase inhibitor deficiency (case 1)
Patient 1 was diagnosed with acquired angioedema caused by C1-esterase inhibitor deficiency (AAE). AAE is rare and manifests as recurrent subcutaneous non-pruritic swelling of the face, larynx, tongue, extremities, abdominal wall, and/or genital organs, without urticaria.3 An attack typically lasts for 24 to 72 hours.3 Angioedema of the tongue and/or larynx can be life-threatening.3 The median age at first presentation is 57-62 years.3,4 In most patients with AAE, C4, C1-esterase inhibitor and C1q levels, and C1-esterase inhibitor activity are reduced.5,6 A majority (75-83%) of patients with AAE also have a lymphoproliferative disorder, and 26-57% of patients with AAE have an ‘MGUS-like’ monoclonal gammopathy.3,4,7 There are two hypotheses on the association between C1-estarase inhibitor deficiency and these lymphoproliferative disorders: (1) the produced monoclonal immunoglobulin has anti-C1-esterase-inhibitor-activity, and (2) excessive activation of C1 by immune complexes containing the monoclonal immunoglobulin results in consumption of C1 inhibitor.8 If detectable, the anti-C1-esterase inhibitor antibodies frequently exhibit the same isotype as the monoclonal immunoglobulin.7
Acute attacks of angioedema can be treated with tranexamic acid (inhibition of plasmin formation; only for moderate attacks), icatibant (bradykinin receptor antagonist) and/or C1-esterase inhibitor concentrate.3 Tranexamic acid and danazol (an androgenic steroid that stimulates hepatic production of C1-esterase inhibitor) are effective in reducing the frequency of angioedema attacks, in 76-93% and 33-75% of patients, respectively.3,4 Moreover, based on limited data, administration of rituximab and treatment of the underlying disease seem also effective in reducing the frequency of attacks.3,7,9 In five of the six patients with MGUS, treatment with rituximab resulted in response (defined as no attacks or > 50% reduction in attacks over the next six months).3
Monoclonal gammopathy of renal significance (cases 2 and 3)
Patients 2 and 3 were diagnosed with monoclonal gammopathy of renal significance (MGRS).
The term MGRS was introduced in 2012 by the International Kidney and Monoclonal Gammopathy Research Group.10 The diagnostic criteria of MGRS are (1) B-cell or plasma cell clonal lymphoproliferation that does not cause tumour complications or meet any current haematological criteria for specific therapy, and (2) kidney biopsy with presence of one or more lesions that are related to the produced monoclonal immunoglobulin.11 MGRS is an umbrella term for different types of kidney damage, mainly caused by deposition of (fragments of) monoclonal immunoglobulins. These different types of MGRS are categorised by localisation of lesions in the nephron and their pathological features (fibrillar, microtubular, inclusion/crystalline deposits, non-organized monoclonal immunoglobulin deposits, or absence of monoclonal immunoglobulin deposits).11 In general, MGRS should be suspected, and a kidney biopsy should be performed in patients with a monoclonal gammopathy and unexplained kidney disease, in patients with known risk factors for chronic kidney disease but an atypical clinical course, and in patients with kidney disease and monoclonal gammopathy aged < 50 years.11
In general, the overall survival of patients with MGRS is significantly better than multiple myeloma, but the renal outcomes are not. Moreover, if left untreated, the recurrence rate after kidney transplantation is very high (> 80%).10,11 Thus, treatment of MGRS is indicated in order to reduce mortality, prevent kidney failure, improve kidney function and, in patients with end-stage renal disease, prevent recurrence after transplantation. The treatment goal of MGRS is complete haematologic remission by targeting the underlying B-cell clone by chemotherapy or an autologous stem cell transplantation.12
Cryoglobulinemia type II (case 2) The patient of case 2 was diagnosed with cryoglobulinemia type II with skin vasculitis and glomerulonephritis secondary to a monoclonal gammopathy. Cryoglobulinemia is defined by the presence of cryoglobulins, immunoglobulins that precipitate at a temperature below 37 ⁰C, in the circulation. Brouet et al. defined three types of cryoglobulinemia in 1974.13 Only type I and type II cryoglobulinemia are associated with monoclonal gammopathies.
The cryoglobulins in type II cryoglobulinemia are immune complexes of a monoclonal immunoglobulin (usually IgM kappa) with antibody activity against IgG and polyclonal IgG.13 As in our patient, serum complement levels are usually decreased in type II cryoglobulinemia.14 Most cases (82%) of type II cryoglobulinemia are associated with a chronic hepatitis C virus infection.14 Other associated conditions are monoclonal gammopathies, haematological malignancies, hepatitis B virus infection, HIV infection, and autoimmune diseases, mainly Sjögren syndrome.14,15 Slightly more than half (53-60%) of patients with type II cryoglobulinemia, including our patient, have vascular purpura, often triggered by standing or exercise.13,14 Other symptoms are Raynaud’s phenomenon, arthralgia and arthritis, as well as peripheral neuropathy and kidney damage (prevalence of the latter is 33-35%).13,14 Type II cryoglobulinemia can also be asymptomatic.14 Treatment is not always indicated. In patients without symptoms or with only few systemic symptoms and/ or only episodic purpuric flares, observation alone is recommended.12 An underlying condition, such as hepatitis C virus infection, should always be treated. Indications for treatment are the presence of vasculitis, progressive systemic symptoms, and MGRS.12 Monotherapy with high-dose corticosteroids is very effective in reducing symptoms of vasculitis.
Treatment with rituximab is indicated in patients with recurrent symptoms or renal involvement.12 Next, chemotherapy (rituximab-containing regimen or bendamustine) should be considered in patients with Waldenström macroglobulinemia or B-cell lymphoma with symptoms more significant than occasional purpura.12 In patients with acute severe systemic symptoms and/or severe organ involvement plasma exchange should be considered.12
CONCLUSION
We have described three cases of an ‘MGUS-like’ monoclonal gammopathy causing significant morbidity. Assessment for presence of monoclonal gammopathy is highly recommended in patients with unexplained kidney damage (MGRS), acquired angioedema due to C1-esterase inhibitor deficiency, and/or unexplained purpura. Asymptomatic monoclonal gammopathy (MGUS) must be distinguished from monoclonal gammopathies with significance, as prognosis and management are vastly different.
DISCLOSURE
There was no funding for this study. There are no conflicts of interest.
REFERENCES