KEYWORDS
Focal segmental glomerulosclerosis, multiple autoimmune disease, polyautoimmunity, Sjögren’s syndrome
INTRODUCTION
Sjögren’s syndrome is a rare autoimmune condition characterised by lymphocytic infiltration of salivary and lacrimal glands associated with anti-SSA (Ro) or anti-SSB (La) autoantibodies. Extra-glandular manifestations are frequently observed and often include renal involvement.1 Sjögren’s syndrome-associated renal disease usually consists of tubulointerstitial nephritis (75% of patients) and glomerulopathy, such as membranous proliferative glomerulonephritis.1-3. Focal segmental glomerular sclerosis (FSGS) as a dominant pathological finding is found rarely in patients with Sjögren’s disease (2/24, 3/103, 0/25 of renal biopsies1-3 and three case reports, respectively).4-6 We present a case with multiple autoimmune phenomena, including Sjögren’s syndrome with a FSGS tip lesion and discuss possible immunological phenomena involved.
CASE REPORT
A 57-year-old non-smoking woman had developed celiac disease and autoimmune hypothyroidism in 1990, and autoimmune gastritis, autoimmune thrombocytopenia, and Sjögren’s syndrome in 2010. Sjögren’s syndrome was diagnosed based on the combination of sicca syndrome, due to chronic sialo-adenitis, arthralgia, and positive anti-extractable nuclear antigen antibodies consisting of anti-SSA (> 240 U/ml) and rheumatoid factor (6.1 IU/ml) with diffuse hypergammaglobulinemia [(immunoglobulin G (IgG) 16.3 g/l (ref: 7.0-16.0 g/l); IgA 5.2 g/l (ref: 0.7-4.0 g/l); IgM 1.16 g/l (ref: 0.4-2.3 g/l)]. Treatment with hydroxychloroquine was discontinued after half a year because of side effects, followed by intramuscular triamcinolone acetonide injections (80 mg every six weeks) with a good result.
One year after cessation of the triamcinolone acetonide treatment in 2017, thrombocytopenia was progressive (33 x 109/l) and she developed a nephrotic syndrome with 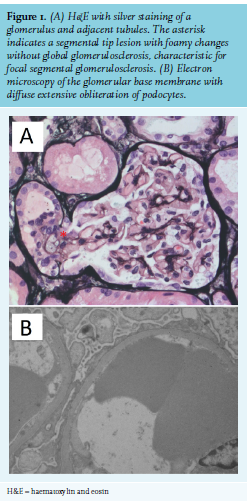 diffuse oedema and a 14 kg weight gain, hypoalbuminemia (albumin 22 g/l), heavy proteinuria (total protein 8.6 g/10 mmol creatinine), and an increase in creatinine from 81 to 105 µmol/l within two weeks. Serum complement levels were normal (C3 1.16 g/l, C4 0.35 g/l) and anti-phospholipase-A2-receptor (anti-Pla2R) was negative. Renal biopsy revealed a tip lesion variant of FSGS with negative immunofluorescence (IgA, IgG, IgM, kappa, lambda, C1q, C3, and Congo red) and no evidence of thrombotic microangiopathy (figure 1). The patient was treated with furosemide, angiotensin-converting enzyme inhibitor perindopril, and prednisolone (1 mg/kg). Within two weeks, the patient lost 12 kg, albumin levels increased to 24 g/l, protein excretion decreased to 1.7 g/10 mmol creatinine, and creatinine normalised. During the tapering of prednisone to 40 mg, protein excretion increased to 2.7 g/10 mmol creatinine. Because of side effects of long-term corticosteroid usage, rituximab was given as a corticoid-sparing agent. Five months later, kidney function normalised (creatinine 62 µmol/l; albumin 37 g/l; urinary total protein 0.09 g/10 mmol creatinine).
diffuse oedema and a 14 kg weight gain, hypoalbuminemia (albumin 22 g/l), heavy proteinuria (total protein 8.6 g/10 mmol creatinine), and an increase in creatinine from 81 to 105 µmol/l within two weeks. Serum complement levels were normal (C3 1.16 g/l, C4 0.35 g/l) and anti-phospholipase-A2-receptor (anti-Pla2R) was negative. Renal biopsy revealed a tip lesion variant of FSGS with negative immunofluorescence (IgA, IgG, IgM, kappa, lambda, C1q, C3, and Congo red) and no evidence of thrombotic microangiopathy (figure 1). The patient was treated with furosemide, angiotensin-converting enzyme inhibitor perindopril, and prednisolone (1 mg/kg). Within two weeks, the patient lost 12 kg, albumin levels increased to 24 g/l, protein excretion decreased to 1.7 g/10 mmol creatinine, and creatinine normalised. During the tapering of prednisone to 40 mg, protein excretion increased to 2.7 g/10 mmol creatinine. Because of side effects of long-term corticosteroid usage, rituximab was given as a corticoid-sparing agent. Five months later, kidney function normalised (creatinine 62 µmol/l; albumin 37 g/l; urinary total protein 0.09 g/10 mmol creatinine).
DISCUSSION
We report a patient with at least five auto-immune phenomena, including Sjögren’s syndrome, who developed a nephrotic syndrome due to a FSGS tip lesion. A FSGS tip lesion often responds better to glucocorticoid therapy than other histological variants of FSGS. Approximately three-quarters of FSGS variants are ‘not otherwise specified’ according to the Colombia classification and reach complete remission in 13-24% after 1-2 years of follow up. The ‘collapsing’ variant (4-12% of cases) is the least favourable, with complete remission rates from 8-14%, followed by the ‘perihilar’ variant, with rates around 10%; the rare ‘cellular’ variant (1-3% of cases) and more common tip lesion (10-14% of cases) achieve remission more often: 33-39% and 50-61%, respectively.7
Although FSGS has rarely been reported in Sjögren’s syndrome1-3 we consider Sjögren’s syndrome as the most likely cause and, to the best of our knowledge, this is the first case report of a FSGS in association with Sjögren’s syndrome that is classified as a tip lesion. In autoimmune hypothyroidism there have been anecdotical reports of FSGS.8 However, since autoimmune hypothyroidism is a common disorder, these might be unrelated phenomena and represent the distribution of this renal pathology in the general population. The other autoimmune phenomena present in our patient – celiac disease, atrophic gastritis, and thrombocytopenia – have not been associated with FSGS.
Although it is not possible to prove a causal link between polyautoimmunity and a FSGS tip lesion with certainty in our patient, both disease entities involve dysregulation of the immune system. The exact cause of FSGS remains unclear, but is thought to involve a glomerular permeability factor. This factor would affect the glomerular capillary wall and thereby lead to podocyte fusion and proteinuria. Additionally, since the monoclonal antibody rituximab agent (that depletes the CD20 B-cell population) appears effective as a corticoidsparing agent in glucocorticoid-sensitive FSGS, B-cells may play a role in the pathophysiology.9 Similarly, in nearly all autoimmune diseases, dysfunctional T-cells play a key role: CD4-positive T-lymphocytes lose immunological tolerance to self-antigens and B-cells produce a spectrum of more or less specific auto-antibodies. Ciclosporin, which mainly inhibits T-cells, has also been successful in treating FSGS in Sjögren’s syndrome in a few cases.4,6 Interestingly, diffuse epithelial foot process effacement has also been described in systemic lupus erythematosus (SLE) without immune complex deposition, occurring more commonly with a disease flare.10 One may speculate that activated T-cells are implicated in the pathogenesis of this so called ‘lupus podocytopathy’.
Since our patient presented with at least five autoimmune phenomena, this may represent more severe dysregulation of the immune system and, although this is highly speculative, leaving her more prone to FSGS. One-third of patients with Sjögren’s syndrome has polyautoimmunity and 9% has three or more autoimmune phenomena, most commonly hypothyroidism.11 Autoimmune diseases are linked to genetic and environmental factors, both may contribute to polyautoimmunity. Since various renal pathology has been reported in Sjögren’s syndrome that have varying prognoses and require different treatment, we consider it essential to perform a renal biopsy to establish the correct diagnosis. In conclusion, polyautoimmunity including Sjögren’s syndrome may be associated with a FSGS tip lesion.
REFERENCES