KEY WORDS 
Post transfusion purpura, HPA-1a, refractoriness, acute leukemia
INTRODUCTION
Post-transfusion purpura (PTP) is a severe transfusion reaction that was first described in 1961.1 The incidence of PTP is estimated at 1:50.000-100.000 transfusions.2 PTP typically occurs after whole blood transfusion in women who have had a first immunization reaction during pregnancy, but also in subjects who have had previous transfusions. PTP is characterized by antibody formation against Human Platelet Antigens (HPA), which are lacking on platelets of the recipient. This nearly always results from immunization against HPA-1a-antigens in HPA-1a-negative subjects but occasionally, immunization against other HPA antigen types occurs.3-5 The prevalence of HPA-1a-negative individuals in the Caucasian population is about 2%.6 Characteristically, PTP presents 5-10 days after transfusion of blood products with deep thrombocytopenia (< 10 x 109/l), fever, shivering from cold and refractoriness to platelet transfusions.7 Most patients recover spontaneously within weeks, but for patients with a secondary high bleeding risk, PTP is associated with a mortality rate of 10-20%.8
The mechanism of PTP is not precisely known. Both the transfused donor platelets and the patient’s autologous platelets are destroyed. This ‘innocent bystander’ effect may be explained by the fact that autologous platelets absorb immune-complexes against allogeneic HPA-antigens through interaction with the Fc-receptor and are subsequently sequestrated in the spleen.1,3,8,9 Another hypothesis suggests the production of platelet autoantibodies next to the HPA-1a alloantibodies.1,9,10 A third hypothesis suggests absorption of soluble HPA-antigens in donor plasma or preservative on autologous platelets, which subsequently react with HPA antibodies.2,5 We describe a suspected PTP in a patient receiving chemotherapy for acute myeloid leukaemia (AML) and discuss the complexity of diagnosis and therapy.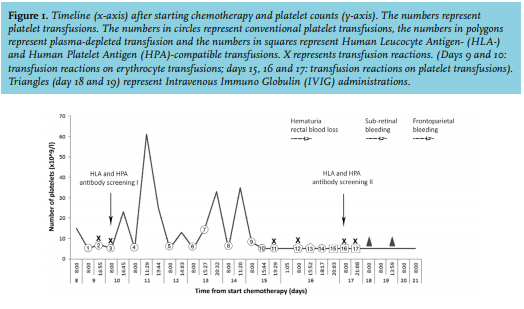
CASE REPORT
A 57-year old woman, mother of two children, presented with AML (Hb: 5.7 g/dl; leukocytes: 139 x 109/l; platelets: 78 x 109/l). During induction chemotherapy, multiple platelet concentrates and red blood cell units were transfused (figure 1). On days 9 and 10, red blood cell infusion was accompanied by fever and shivering. Clemastine and prednisolone were administered which reduced these symptoms. Therefore, preceding all subsequent transfusions, clemastine and prednisolone were administered. While platelet transfusions 1-3 were administered without a transfusion reaction, the corrected count increments (CCI) after one and 24 hours (CCI-1 and CCI-24) were zero. Therefore, Human Leukocyte Antigen (HLA) and HPA antibody screening and genotyping were performed. Genotyping showed that the patient was negative for the HPA-1a antigen and no HLA nor HPA antibodies were found. Furthermore, imaging ruled out splenomegaly. Platelet transfusions 4, 7 and 8 resulted in a CCI-1 > 2.5, however, platelet counts decreased to < 5.0 x 109/l within 24 hours, resulting in nose bleeds and hematomas. After platelet transfusion 11, the platelet count remained < 5.0 x 109/l, and transfusion reactions no longer responded to administration of clemastine and prednisolone. Febrile reactions were observed only after transfusions. There was no sign of infection. The plasma solution of the platelet concentrates 12 and 13 was substituted by platelet additive solution in an attempt to prevent transfusion reactions. But both transfusions resulted in a CCI-1 of zero. At this moment, platelet transfusions were mandatory because of hematuria and rectal bleeding. Because of suspicion of an immunological cause, HLA and HPA antibody screening was repeated on day 17 and HPA-1a and HLA-matched donor platelet concentrates were transfused; however, no increase in platelet count was observed (transfusions 15-17). On day 18, the patient developed a sub-retinal bleeding. At that moment, the second antibody analysis revealed strong reactive HPA-1a antibodies and multiple HLA antibodies. Therefore, PTP was suspected and transfusions were stopped and administration of intravenous immunoglobulins (IVIG) was started. Two days after starting IVIG, the patient developed a lethal frontoparietal bleeding due to persisting thrombocytopenia.
DISCUSSION
Since thrombocytopenia has several causes, finding the right diagnosis may be challenging. During AML treatment, thrombocytopenia most commonly is disease or therapy-related. Therefore, diagnosing PTP can be difficult and delayed.
The corrected count increment after one hour (CCI-1) is an important tool to discriminate between immunological (CCI-1 < 7.5) and non-immunological (CCI-1 > 7.5) causes for platelet transfusion refractoriness. In case of an immunological cause, specification of HLA or HPA alloantibodies and/or platelet glycoprotein reactive antibodies can provide valuable information on the cause. Here, despite decreased CCI-1 values, no HLA and HPA antibodies were found in the first analysis. It appears that, especially in case of an HPA-1a-negative patient, reanalysis of antibody screening should be considered when an immunological cause of thrombocytopenia is clinically suspected, because increase of HLA or HPA antibody titers may be delayed and result into a negative laboratory test.
Thrombocytopenia in AML is common and can mostly be attributed to chemotherapy, inadequate response to platelet transfusions, sepsis and HLA or (rarely) HPA alloimmune antibodies. Furthermore, the differential diagnosis includes heparin-induced thrombocytopenia, disseminated intravascular coagulation (DIC), thrombotic thrombocytopenic purpura (TTP) and medicationinduced thrombocytopenia. Apart from chemotherapy, this patient did not use any medication known to cause thrombocytopenia, neither did she receive heparin. No signs of sepsis or infection were observed. DIC and TTP were ruled out by normal laboratory findings for activated Partial Thromboplastin Time, Prothrombin Time, fibrinogen and absence of fragmentocytes. Although chemotherapy definitely contributes to thrombocytopenia, here we could discriminate HPA-1a type antibodies, which is very suggestive for PTP in HPA1a-negative individuals. In case of PTP, prophylactic platelet transfusions, even compatible with HPA and HLA antigens, are contra-indicated.11 IVIG is the primary choice of treatment, since it has response rates of 75-95% and a rapid onset of action.11,12 Alternatively, steroids may be administrated together with IVIG, although responses appear unpredictable.13 Plasmapheresis, which is occasionally considered as a second line treatment, results in improved platelet counts within 2-4 days in 80% of patient cases.14,15 Unfortunately, in our patient, administration of IVIG and corticosteroids did not result in an increase of the platelet count and plasmapheresis could not be applied because rapid deterioration led to her demise.
CONCLUSION
PTP, in particular in patients treated for AML, may not be easily recognized. This delay may be fatal as is illustrated in our patient. When clinically suspected, PTP should not be excluded in platelet transfusion refractory HPA-1a-negative patients without detectable HPA-1a antibodies. However, even with timely recognition, PTP has a high mortality rate, especially in high-risk patients, like our patient with AML.
DISCLOSURES
All authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES