KEYWORDS
C3 glomerulonephritis, C3 glomerulopathy, center of expertise, dense deposit disease, membranoproliferative glomerulonephritis
INTRODUCTION
C3 glomerulopathy is a rare renal disease that has been distinguished for about 10 years, when its pathophysiology was discovered to differ from other types of membrano-proliferative glomerulonephritis.1,2 C3 glomerulopathy appears to be caused by deposition of complement factor C3 without other complement factors or immunoglobulins in the glomeruli, as explained hereafter. Knowledge and comprehension of this disease are rapidly evolving. According to the Dutch National Plan on Rare Diseases, centers of expertise are allocated by the Ministry of Health in consultation with the European organisation for the care for rare diseases, Orphanet and the Dutch organisation for patients with rare diseases (Vereniging Samenwerkende Ouder-en Patiëntenorganisaties, VSOP). The national centers of expertise strive to collaborate within a European Reference Network. Because C3 glomerulopathy is a rare and new disease, clinical and scientific expertise are concentrated in such centers. Radboud University Medical Center (Radboudumc) and Leiden University Medical Center (LUMC) are centers of expertise on C3 glomerulopathy in the Netherlands. This review provides a succinct overview of the current diagnosis and treatment of C3 glomerulopathy at LUMC.
EPIDEMIOLOGY
The incidence of C3 glomerulopathy has been estimated at 1-2 per million per year.3 It is diagnosed in 1-2% of kidney biopsies.3,4 C3 glomerulopathy typically occurs in childhood or adolescence, but may also be diagnosed at older ages due to slow disease progression.3-14 It occurs equally in men and women,3,5-7,10-12 predominantly in Caucasian ethnicities,5,8,9,13,15 and sometimes with a familial distribution.3,6,7,12,16 It commonly presents after an infection, which is thought to be a precipitating or eliciting risk factor, as in other glomerulonephritides.13,14,16-18 No specific types of infections have been associated with C3 glomerulopathy, although respiratory tract infections seem most common.3,5,7 Other possible risk factors include vaccination, immunosuppressive, cytotoxic, and contraconceptive medication, pregnancy and childbirth.17
PATHOPHYSIOLOGY
C3 glomerulopathy is caused by excessive activation of the alternative complement pathway.16,17,19-22 The complement pathways comprise circulating factors that sequentially activate each other in order to eliminate pathogens and damaged cells, and to guide and regulate immune cells and antibodies. Extensive reviews of the complement pathways are available elsewhere;11,17,20-24 a brief overview is given here and in figure 1. The alternative pathway is continuously active at a low level. Complement factor C3 is cleaved into C3a and C3b. C3a is an anaphylatoxin with proinflammatory and procoagulant effects. C3b binds factor Bb, after which, the complex C3bBb amplifies the pathway by cleaving additional C3 and creating more C3b and C3bBb. Because of this action, C3bBb is called C3 convertase.17,20-24 Large amounts of C3a and C3b can be formed, since C3 is abundant in plasma, accounting for approximately 2% of all proteins.9 Because of its continuous spontaneous activity, regulatory factors suppress the activity of the alternative pathway in order to terminate its effects after the elimination of a pathogen or damaged cell, and to avoid harmful effects to other cells. C3 glomerulopathy may arise when a mutation renders such a factor deficient or dysfunctional; this has been shown in animal models and also observed in families with inherited C3 glomerulopathy.16,20 Most mutations affect circulating factor H. Mutations of thrombomodulin – known for its role in coagulation – can enhance the activity of the suppressing factor I. Mutations of C3 can make it resistant to a suppressing regulatory factor.10,20 Other regulatory factors enhance the activity of the alternative pathway. C3 glomerulopathy may arise when a mutation renders factor B, factor H-related factor (CFHR) or properdin hyper-functional or increases the susceptibility of C3 to one of these enhancing regulatory factors.6,7,16,17,20 Excessive activation of the alternative pathway may also be caused by autoantibodies. An autoantibody that binds and stabilizes C3 convertase is called C3 nephritic factor. Some C3 nephritic factors only bind C3 convertase in the presence of properdin. Other autoantibodies bind and eliminate suppressing regulatory factors like factor H, or bind and stabilize enhancing regulatory factors like factor B.7,16,17,20 The classical pathway and the lectin pathway can activate the alternative pathway. These pathways are activated by antibodies, immune complexes, or components of pathogens or damaged cells. Their activation results in the formation of the C4bC2a complex, which cleaves C3 and creates more C3b and C3bBb. C4bC2a is therefore also called C3 convertase of the classical pathway.17,21,23,24 Autoantibodies that bind and stabilize this C3 convertase are called C4 nephritic factor.11,25 The alternative, classical and lectin pathway all initiate the terminal pathway by forming C5 convertase, which cleaves C5 into C5a and C5b. C5a acts as an anaphylatoxin like C3a. C5b binds C6, C7, C8 and multiple copies of C9, forming C5b-C9, also called C5b-9 or the membrane attack complex, since it forms a borehole in the membrane of a pathogen or cell and causes its lysis.11,17,20-24 Autoantibodies that bind and stabilize C5 convertase are called C5 nephritic factor.11 Confusingly, properdin-dependent C3 nephritic factor is sometimes also called C5 nephritic factor, as it is related to excessive activation of the terminal pathway. A mutation or autoantibody alone is insufficient to cause C3 glomerulopathy. They occur in other kidney diseases, as well as in apparently healthy persons, some in up to 8% of the general population. Multiple mutations, autoantibodies or additional genetic and environmental risk factors that activate complement factors – such as an infection – are thought to be required to elicit C3 glomerulopathy.7,10,17,21,26 Despite these insights into its pathophysiology, the predisposing and/or eliciting causes of C3 glomerulopathy remain unknown in many patients, more often so in adults than children.13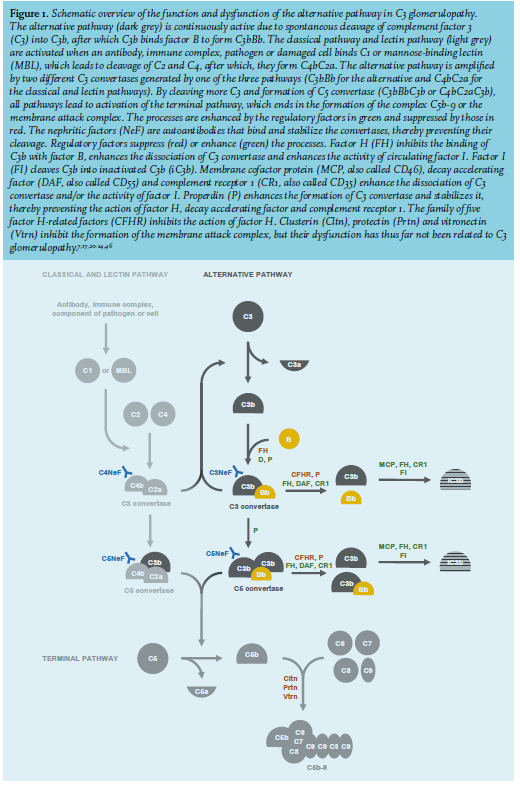
CLINICAL PRESENTATION
The presentation of C3 glomerulopathy varies widely. It can present with signs of a nephritic syndrome, including proteinuria (90-95%), microscopic (64-88%) or macroscopic hematuria (16-38%), renal impairment (14-59%) and hypertension (21-46%).3-8,10-12,14,18,27,28 A nephrotic syndrome is found in 27-55%.5,6,10-13,27,28 Reports of the duration of symptoms until diagnosis differ between less than a year and multiple years.8,10,12 Children usually have a milder and slower course of disease than adults.13,29 For all patients suspected of C3 glomerulopathy – and likewise any glomerular disease – we evaluate the presence of these symptoms, general symptoms of autoimmune disease and the risk factors mentioned before (table 1). In up to one-third of adult patients with C3 glomerulopathy, a monoclonal gammopathy is found, with increasing numbers at older ages.4,5,11,13,14,30-32 The gammopathy most commonly occurs without a lymphoproliferative malignancy,5,14,30-33 which would previously have been considered a monoclonal gammopathy of undetermined significance, but is now, in the presence of renal disease, called a monoclonal gammopathy of renal significance. Among patients aged 50 years and older, the gammopathy is a monoclonal gammopathy of renal significance in 44-83%, a multiple myeloma in 44% and another malignancy in 11-17%, all without cast nephropathy or glomerular deposition of immunoglobulins or light chains.4,30,34 The paraprotein is thought to act as an autoantibody that inhibits regulatory factors, stabilizes C3 convertase like C3 nephritic factor, or cleaves C3 and consequently activates the alternative pathway.4,17,30-32,34,35 All patients should be tested for the presence of monoclonal gammopathy using protein electrophoresis.19 Ocular drusen and acquired partial lipodystrophy are associated with C3 glomerulopathy. Ocular drusen are deposits of complement factors between the retinal epithelium and the underlying Bruch’s membrane. Drusen resemble the deposits of complement factors in the glomeruli, discussed hereafter, and are a result of the structural and functional similarities between the Bruch’s membrane and the glomerular basement membrane. Drusen are also found in age-related macular degeneration, but occur in younger C3 glomerulopathy patients. They can lead to visual impairment.5,7,36-39 We refer patients to an ophthalmologist to be screened for drusen by fluorescein angiography and optical coherence tomography. Even in the absence of symptoms, screening for drusen may be helpful for timely prevention or treatment and for the diagnosis of C3 glomerulopathy. Acquired partial lipodystrophy involves the loss of subcutaneous fat from the face, neck, arms and trunk. Fat tissue is destructed by complement factors, perhaps because adipocytes themselves produce C3, factor B and C3 convertase.5,6,7,37,40 We examine patients for lipodystrophy. 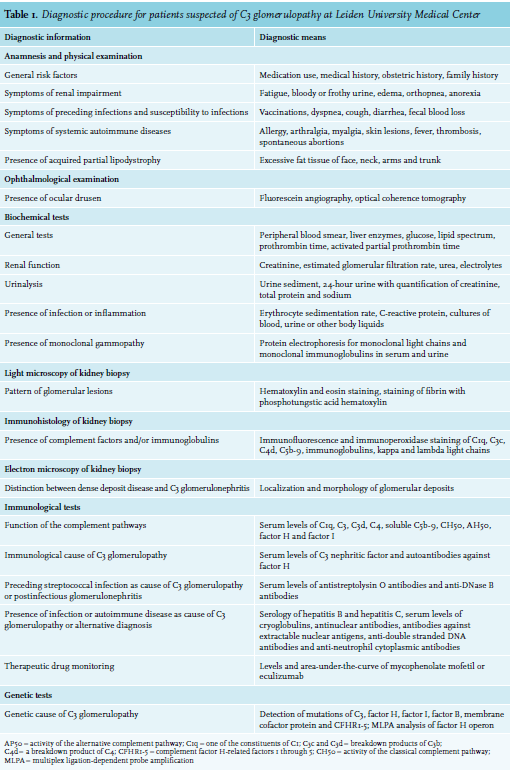
HISTOLOGICAL PATTERNS
The excessive activation of the alternative complement pathway that characterizes C3 glomerulopathy results in deposition of complement factor C3 in glomeruli. Although the relation between histological patterns and clinical presentation remains unclear,37 a kidney biopsy is required to evaluate the presence, localization, and composition of the depositions and to confirm the diagnosis.41 Furthermore, a kidney biopsy is the only method for distinguishing the two types of C3 glomerulopathy: dense deposit disease and C3 glomerulonephritis. Their differences in pathophysiology remain unclear,7,26 since neither their genetic causes6,9,10,26 nor clinical presentations differ, except that dense deposit disease is uncommonly seen in the presence of a monoclonal gammopathy and confers a lower risk of renal impairment.3,6,7,10,12,13 Under light microscopy, C3 glomerulopathy presents as membranoproliferative glomerulonephritis in 44-76% of patients, mesangial proliferative glomerulonephritis in 21-28%, diffuse endocapillary proliferative glomerulonephritis in 8-19% and crescentic glomerulonephritis in 9%. Dense deposit disease presents more often with acute crescentic lesions, while C3 glomerulonephritis presents more often with chronic fibrotic and sclerotic lesions,3-6,10,12,27,28 although, confusingly, the opposite has also been reported.13 Children present less commonly with glomerular and interstitial fibrosis than adults.5,29 Immunohistology is used to identify the constituents of the glomerular deposits. Deposits of C3 and its breakdown products define C3 glomerulopathy,19 while deposits of C1 and C4 are scarce or absent.42 We have not routinely evaluated the presence of other complement factors. Deposits of C4 may be associated with monoclonal gammopathy.43 Deposits of C5b-9 are thought to reflect activation of the terminal pathway, but can also be found in apparently healthy kidneys and after kidney transplantation, particularly in vascular and fibrotic lesions, suggesting a role in tissue damage or tissue repair.5,19 Deposits of immunoglobulins occur in variable amounts and with uncertain relevance.8,19 Deposits of immunoglobulins are most common of the IgM type3-5,12 and are located subendothelially or subepithelially.4 In addition to immunoglobulins, we evaluate the presence of light chains to unveil possible monoclonal gammopathy as an underlying cause of C3 glomerulopathy.37 The presence of immunoglobulins or light chains may remain masked in routinely-used freshly frozen samples, and revealed only in formalin-fixed paraffin-embedded samples to which a protease has been added.43,44 The latter technique should therefore be considered, especially when a monoclonal gammopathy is not detected in the freshly frozen sample but suspected, for example, due to deposits of C4.37 Studies of single glomeruli using mass spectrometry have confirmed that the deposits in C3 glomerulopathy consist of C3 and its breakdown products, together with C5b-9 and uncommonly C4, but not of immunoglobulins.30,34,42,45 By agreement, C3 glomerulopathy has been defined as a glomerulonephritis with dominant staining for C3 that is at least two orders of magnitude more intense than other complement factors, immunoglobulins or immune complexes.19 More strict definitions, excluding patients with any deposits of immunoglobulins, miss many probable cases of C3 glomerulopathy, although even the current definition may miss such cases.8,19 Electron microscopy is used to localize the glomerular deposits.37 In dense deposit disease, by definition, the deposits are electron-dense and located within the glomerular basement membrane. C3 glomerulonephritis is characterized by less dense and less concrete granular deposits in the glomerular basement membrane3,7,10,12,19 and deposits in the mesangium, the subendothelial or subepithelial space.3,5,7,10,12,19 These deposits are found less often in dense deposit disease in addition to the electron-dense deposits in the glomerular basement membrane. Deposits are uncommonly found in Bowman’s capsule and the tubular basement membrane.3,19
TEST OF THE COMPLEMENT PATHWAYS
Several tests of the complement pathways should be performed in all patients suspected of C3 glomerulopathy. Consistent with activation of the alternative pathway and glomerular deposition of C3, the serum level of C3 is reduced in the majority (41-76%), and the serum level of C4 is reduced in only a minority (1-23%) of patients.3,5-14,27,28,37 The level of C3 is more often reduced in children (83-100%) than adults (41-57%).5,13 Subsequent activation of the terminal pathway is reflected by a low level of C5 and an elevated level of circulating C5b-9.9,11,26 Unlike C3 and C4 levels, we have not measured the levels of C5 and C5b-9 routinely. The measurement of circulating C5b-9 is difficult, as it requires appropriate sampling, handling and storage of plasma to prevent iatrogenic activation of the complement pathways. Helping to distinguish between both types, the level of C3 appears more often reduced in dense deposit disease3,6,8,10,12 and in patients with C3 nephritic factor.6 The level of C5b-9 appears higher in patients with C3 glomerulonephritis and in patients with a properdin-dependent C3 nephritic factor.9,11,12,21,26,46 These results suggest that the alternative pathway is activated more in the former category of patients and that the terminal pathway is activated more in the latter category of patients. We measure the activity of the complement pathways as the hemolytic activity of a patient’s serum directed against animal erythrocytes. The so-called CH50 represents the activity of the classical pathway, while the AH50 or AP50 represents the activity of the alternative pathway. We measure C3 nephritic factor and autoantibodies against factor H as potential causes of C3 glomerulopathy. C3 nephritic factor is present in 44-78% of patients, and more often in dense deposit disease (78-86%) than C3 glomerulonephritis (38-59%),5-7,9-12,14,17,20,25 and more often in children (91-100%) than adults (33-46%).29 Other types of nephritic factor are less common. Different types of nephritic factor can also be present simultaneously.11,25 Tests of C3 nephritic factor have not been standardized and vary between laboratories. Some laboratories run different tests in parallel. We make use of the tests at Sanquin or Radboudumc47 in the Netherlands or the University of Iowa, United States48 and repeat a test elsewhere if its result is uncertain. The tests are currently evaluated to enhance their reliability, to understand their comparability and to distinguish different types of nephritic factors. We incidentally measure other autoantibodies, like C4 nephritic factor or antibodies against factor B, if no other cause of C3 glomerulopathy has been found and if the presence of autoantibodies is suspected, for example, based on the presence of antinuclear antibodies. We do not rely on other tests of the complement pathways, since these measurements can be performed in only a few laboratories and have not been validated. As part of the national COMBAT Consortium (COMplement: Basis mechanisms, Assay development and novel Therapy), we currently evaluate and implement novel tests.
GENETIC TESTS
All patients require referral to a clinical geneticist to test for mutations, copy number variation, hybrid genes and other genetic rearrangements in the genes of complement factors, most specifically C3, factor B, factor H, factor I and CFHR. The processing time and the reliability of these tests are improving. Several additional alleles that confer an elevated risk of C3 glomerulopathy are only tested for research purposes.6,9,10
DIFFERENTIAL DIAGNOSIS
The aforementioned tests help to distinguish C3 glomerulopathy from other types of glomerulonephritides that may closely resemble C3 glomerulopathy and are also caused by excessive activation of complement factors, such as immune complex-mediated glomerulonephritis and post-infectious glomerulonephritis.2,6-8,10,12,19,49,50 Still, a distinction is often very difficult. Autoantibodies and genetic mutations do not consistently differ between these three types of glomerulonephritides.6,10 The clinical presentations are similar, except for a nephrotic syndrome occurring more often in immune complex-mediated glomerulonephritis than C3 glomerulopathy.6-8,10,49 In a kidney biopsy, deposits of C4 and its breakdown products, resulting from activation of the classical pathway, are thought to distinguish post-infectious glomerulonephritis and immune complex-mediated glomerulonephritis from C3 glomerulopathy,43,45 but not all studies have confirmed this distinction.37,44 In contrast with C3 glomerulopathy, the deposits in immune complexmediated glomerulonephritis consist less commonly of C3 and C5b-9, more commonly of C1 and C4 and always of immunoglobulins.10,45,49 Subepithelial hump-like deposits as observed with electron microscopy have been thought to characterize post-infectious glomerulonephritis,43 but have also been reported in C3 glomerulopathy, both with and without a preceding infection.3,14,19,37,49 The serum levels of C3, C4, C5b-9 and C3 nephritic factor are similar.6,8,10 Post-infectious glomerulonephritis can sometimes only be discerned by its typical spontaneous resolution in six to twelve weeks. These three glomerulonephritides can even be found consecutively during the course of the disease, before and after kidney transplantation or in different patients within the same family.17 Whereas C3 glomerulopathy arises through excessive activation of the alternative pathway due to dysfunction of circulating complement factors, dysfunction of complement factors at the surface of cells can also lead to excessive activation of the alternative pathway, but this manifests as different diseases, such as atypical hemolytic uremic syndrome. Dysfunction of factors that can circulate and bind the membrane of a pathogen or damaged cell – including C3b, C3 convertase, and factor H – may give rise to either disease. Since the glomerular basement membrane lacks membrane-bound regulatory factors and easily captures circulating complement factors, it is dependent on circulating regulatory factors and affected in C3 glomerulopathy.17,21-23
TREATMENT
No treatment has proven effective and beneficial for C3 glomerulopathy. Recommendations have only been deducted from case series and observational studies7,16,17 and are mostly based on expert opinion.19,37 As a consequence, its treatment has not been standardized and is concentrated in centers of expertise. All patients diagnosed with C3 glomerulopathy should be treated with renoprotective measures, including lifestyle advice, an angiotensin-converting enzyme inhibitor or angiotensin-receptor blocker to control hypertension and proteinuria, and lipid-lowering treatment.17,37,51 Such medication alone has not been shown to protect against progression to end-stage renal disease,6,10 but may improve the protective effect of immunosuppressive medication.5 When a monoclonal gammopathy is suspected to be an underlying cause of C3 glomerulopathy, the patient is referred to a hematologist. Its treatment is thought to be effective in patients with C3 glomerulopathy.4,17,30 In a retrospective study of 50 patients with C3 glomerulopathy and various types of monoclonal gammopathy, renal function and proteinuria improved more often with chemotherapy (74%) than with renoprotective or immunosuppressive medication (5%), and more often if the gammopathy was reduced (83%) than if it was not reduced (28%). The hazard of end-stage renal disease was 83% lower if the gammopathy was reduced.31 In a similar study of 33 patients, improvement of renal function and proteinuria was similar with (38%) and without chemotherapy (41%), and these improvements were seen more often if the gammopathy was reduced (40%) than if it was not reduced (0%).32 Chemotherapy is preferentially used to treat gammopathy due to lymphoproliferative malignancies, but the association between a reduction in gammopathy and an improvement of renal function suggests that chemotherapy may also be considered for the treatment of monoclonal gammopathy of renal significance.29,31,35 If moderate inflammation is present in the kidney biopsy and proteinuria persists despite supportive treatment, or if renal function decreases, then prednisolone and/ or mycophenolate mofetil are recommended.17,37 Neither immunosuppressive medication in general3,6,10,27,28 nor corticosteroids in particular3,5,10 have conferred consistent beneficial effects across various studies. According to two observational studies, mycophenolate mofetil – alone or in combination with prednisolone – offers better protection against renal impairment compared to prednisolone, cyclophosphamide, tacrolimus, cyclosporine or rituximab 27,52 although this was not confirmed in a third study.28 If, despite treatment with prednisolone and mycophenolate mofetil, severe inflammation is present in the kidney biopsy and proteinuria persists, or renal function decreases, then methylprednisolone, cyclophosphamide, tacrolimus or rituximab can be considered, although a beneficial effect has not been confirmed.10,16,17,37 Despite this uncertainty, we prefer rituximab if autoantibodies are present. Exceptional off-label use of eculizumab, a monoclonal antibody that inactivates C5, has been suggested for patients with C3 glomerulopathy.16,17,20,24 Its effects have not been studied in randomized trials. Case reports have described that it improves renal function and proteinuria.7,16,17,20,53 A retrospective study of 26 patients treated with eculizumab reported improvement of renal function and proteinuria in 46% and reduction of histological lesions in 82%. These effects were more often obtained in those with a rapidly progressive course of disease and extra-capillary proliferation, without differences between children and adults.29 In the only prospective study performed in six patients, eculizumab improved renal function in two patients, partially resolved a nephrotic syndrome in a third; it also reduced histological lesions in the two patients with improved renal function and one other patient. Elevated levels of circulating C5b-9 were reduced, but deposits of C5b-9 in the kidney biopsies remained.15,54 Since eculizumab inhibits the terminal pathway, it is expected to benefit particularly those patients with activation of the terminal pathway,15,53,55 reflected by a high amount of C5b-9 in the circulation and kidney biopsy,17,20 which is more common in those with C3 glomerulonephritis.9,10,12,26 Various other complement inhibitors are currently being developed, including inhibitors of C3, inhibitors of C5a and soluble inhibitory regulatory factors.17,20,21,24,56 Treatment with immunosuppressive medication or eculizumab confers a risk of infections, for which antibiotic prophylaxis, vaccination and cautious administration during active infection are indicated.24,37,56 We vaccinate against Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae. Eculizumab has no other known side effects.15,29 If renal function is acutely impaired, especially if crescentic lesions are present in the kidney biopsy, plasmapheresis can be considered in addition to immunosuppressive medication.17,37 Plasmapheresis is expected to be beneficial in cases of dysfunction or deficiency of complement factors or regulatory factors, although this has only been demonstrated in some case reports.7,17 Kidney transplantation is performed in more than half of patients who have reached end-stage renal disease.3 Unfortunately, C3 glomerulopathy often recurs after kidney transplantation, histologically in 67-90% and clinically in 50-60% of patients.3,6,20,31,57 It recurs more often in dense deposit disease than C3 glomerulonephritis3 and in patients with a mutation of a complement factor,17 with rapidly progressive disease or with monoclonal gammopathy.37,57 The transplant is lost due to recurrence in about 50% of patients,3,57,58 which is a large proportion as compared with other glomerulonephritides. The median time of survival of the transplants is around five years; less than a third has survived after 10 years.3,57,58
PROGNOSIS
With the aforementioned treatments, clinical remission of C3 glomerulopathy is reached in 55-62%, with complete remission in 22-32% after two to four years,27,28 or in 35% after six years, with complete remission in 13% of patients.13 Chronic treatment with immunosuppressive medication is necessary in half of the patients to sustain remission.27,28,52 Remission is more often achieved in C3 glomerulonephritis than dense deposit disease.13 Spontaneous remission occurs in rare instances. C3 glomerulopathy leads to end-stage renal disease in 11-52% of patients in, on average, six to ten years.3,5,6,10-14,18 Risk factors of end-stage disease are older age,3-6,11,13,28 renal impairment,3,5-7,13,28 heavy proteinuria at presentation,28 little reduced serum levels of C3 and C5b-9,12 glomerular sclerosis or crescents,3,5,10,12,13,28 interstitial fibrosis or tubular atrophy,13 glomerular deposition of C3 without immunoglobulins,13 monoclonal gammopathy4,30-34 and the absence of C3 nephritic factor or mutations of complement factors.10 Both a nephrotic10 and nephritic syndrome27 have been associated with a worse prognosis. Unfortunately, in clinical practice, neither the presentation nor the results of the aforementioned tests can predict the course of the disease or the effects of treatment.7,37,52
ACKNOWLEGDEMENTS
The description of the histological patterns of C3 glomerulopathy was reviewed by dr. Ingeborg M. Bajema, Department of Pathology, Leiden University Medical Center, Leiden, the Netherlands. Research on C3 glomerulopathy at LUMC is conducted as part of the COMplement: Basic mechanisms, Assay development and novel Therapy Consortium (COMBAT Consortium), funded by the Dutch Kidney Foundation. This review was written without funding.
DISCLOSURES
The authors have no funding or conflicts of interests to disclose.
REFERENCES