KEY WORDS
Hypereosinophilia, parasitic infections, strongyloïdiasis, allergy, hypereosinophilic syndrome, hypereosinophilia of unknown significance, hematological malignancy, chronic eosinophilic leukemia, imatinib, mepolizumab, corticosteroids, ivermectin
CASE HISTORIES
Patient A
A 29-year-old man with an unremarkable past medical history was clinically observed with fever (40°C) and myalgia two weeks after his return from Thailand. He suffered from nausea and mild abdominal pain. During his stay he had a short-lived episode of diarrhea. On physical examination, he had a temperature of 38.6°C and an erythematous maculopapular rash on his right flank and buttocks. Laboratory examination was unremarkable apart from an increased C-reactive protein (CRP) level (223 mg/l). Differential diagnosis included Rickettsiosis (scrub typhus), Salmonella typhi and Melioidosis (Burkholderia pseudomallei). Malaria was excluded. Dengue, Chikungunya and Leptospirosis were less likely based on their shorter incubation periods. During his admission, he was treated with doxycycline and discharged after four days. On follow-up three weeks later, CRP-level had normalized, but a significant leucocytosis of 24 x 109/l had developed with 71% eosinophilia; his absolute eosinophil count was 17 x 109/l. Serological examination and a Triple Faeces Test (TFT) confirmed an infestation with Strongyloides stercoralis. The patient was treated with ivermectin (200 µg/kg) and made a full recovery. His absolute eosinophil count dropped to 0.78 x109/l. Most likely, the patient was infected while walking barefoot along the shores of the River Kwai. These dermatological phenomena due to strongyloïdes is known as larva currens.
Patient B
A previously healthy 62-year-old female presented with diplopia, progressive gait problems and numbness and tingling of hands and feet. She reported a recent flu-like episode with persistent complaints of weight loss and discomfort between the shoulder blades. Physical examination was unremarkable apart from multiple small cervical lymph nodes. Laboratory examinations showed an erythrocyte sedimentation rate (ESR) of 72 mm/hour, leukocytosis of 14 x109/l and CRP of 71 mg/l. Leukocyte differentiation showed 21% eosinophils, with no increase in progenitor cells or basophils. The absolute eosinophil count was 4.4 x109/l. Differential diagnosis included paraneoplastic syndrome associated with underlying malignancy or vasculitis. Guillain-Barre syndrome was found to be unlikely. Full body computerized tomography (CT) scan showed no primary tumor or distant metastasis and magnetic resonance imaging (MRI) showed no intracranial abnormalities or signs of vasculitis. Spinal fluid was normal. Immunological assays to autoantibodies antinuclear antibody and antineutrophil cytoplasmic antibody (ANA/ANCA) were negative and therefore, autoimmune disease was unlikely. Mastocytosis was also unlikely due to normal tryptase levels. During admission, a progressive neurologic condition evolved, which was characterized by neuropathy, gait function disorders, facial paralysis and ptosis. Based on the progressive course of the disease, in combination with hypereosinophilia, a hypereosinophilic syndrome (HES) was suspected. Bone marrow examination revealed an eosinophil count of 20% with no increase in blasts. Bone marrow biopsy showed neither morphological aberrations, nor increase of mast cells. Molecular tests were unremarkable (no C-KIT, BCR-ABL, JAK2 or CALr mutations) and fluorescence in situ hybridization (FISH) did not detect the FIP1L1-PDGFRA fusion gene. Additional cytogenetic tests ultimately revealed a 20q-deletion, as observed in patients with chronic eosinophilic leukaemia (CEL). The patient was treated with high-dose intravenous prednisolone of 60 mg, once daily. Because of the severity of symptoms, she was concomitantly treated with interferon-alpha. She slowly recovered and during several outpatient visits, her eosinophil count eventually normalized.
Patient C
A man, aged 23, known with type I diabetes mellitus presented to the emergency department with acute diarrhea and vomiting. Apart from mild discomfort in the right lower quadrant of his abdomen, physical examination on admission was unremarkable. Laboratory examination showed a leucocytosis of 14 x 109/l. Ultrasound and subsequent CT scan showed ascites with diffuse bowel thickening of his ascending and transverse colon. Follow-up laboratory tests showed the development of a significant eosinophilia with an eosinophil percentage of 48% and an absolute eosinophil count of 7.2 x 109. Detailed history regarding allergies, medication and travel history was inconclusive. Stool sample cultures for pathogens remained negative, as well as serological testing and TFT for parasites. Autoimmune disorders and systemic mastocytosis were disproved by normal immunoglobulins, ANA/ANCA and tryptase. With the working diagnosis of eosinophilic gastroenteritis, a colonoscopy was performed, however this appeared normal and histological biopsy showed no significant eosinophilia or granulomas. Bone marrow examination was performed, but showed no clues of an underlying hematological malignancy (negative tests for FIP1L1-PDGFRA fusion or t(5q32)/ PDGFRB). There were no signs of end-organ damage. Therefore, the patient was diagnosed with ‘hypereosinophilia of unknown significance’. A wait-and-see policy was followed and after two months, he made a full clinical recovery. Laboratory results showed complete normalization of eosinophil counts.
Background
Normal peripher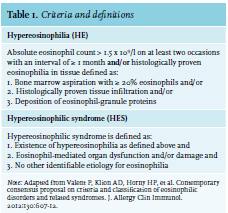 al eosinophil counts vary between 0.04-0.60 x 109/l. Peripheral blood eosinophilia can be divided into mild (0.5-1.5 x 109/l), moderate (1.5-5 x 109/l), and severe (> 5 x 109/l).1 The term hypereosinophilia (HE), as used in this manuscript, is defined as an absolute eosinophil count of > 1.5 x 109/l measured on at least two occasions with an interval of ≥ one month and/or histologically proven tissue infiltration by eosinophils (table 1) 1 Milder eosinophilia can be caused by countless conditions, infectious, autoimmune or cryptogenic notably, HIV infection, granulomatous disorders, polyarthritis nodosa and primary biliary cirrhosis. Furthermore, the term hypereosinophilic syndrome (HES) is defined as the existence of HE in combination with eosinophil-mediated organ damage. The term HES cannot be used if the clinical symptoms and organ involvement have a clear identifiable etiology. The exact incidence and prevalence of HE and HES remain uncertain. The age-adjusted incidence rate is estimated 0.036 per 100,000 individuals per year.2,3
al eosinophil counts vary between 0.04-0.60 x 109/l. Peripheral blood eosinophilia can be divided into mild (0.5-1.5 x 109/l), moderate (1.5-5 x 109/l), and severe (> 5 x 109/l).1 The term hypereosinophilia (HE), as used in this manuscript, is defined as an absolute eosinophil count of > 1.5 x 109/l measured on at least two occasions with an interval of ≥ one month and/or histologically proven tissue infiltration by eosinophils (table 1) 1 Milder eosinophilia can be caused by countless conditions, infectious, autoimmune or cryptogenic notably, HIV infection, granulomatous disorders, polyarthritis nodosa and primary biliary cirrhosis. Furthermore, the term hypereosinophilic syndrome (HES) is defined as the existence of HE in combination with eosinophil-mediated organ damage. The term HES cannot be used if the clinical symptoms and organ involvement have a clear identifiable etiology. The exact incidence and prevalence of HE and HES remain uncertain. The age-adjusted incidence rate is estimated 0.036 per 100,000 individuals per year.2,3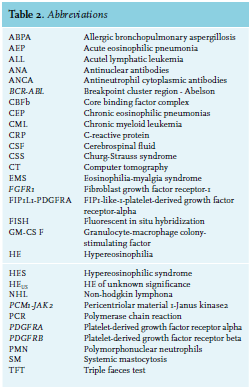
Pathophysiology
Eosinophils are leucocytes originating from CD34+ hematopoietic precursor cells in the bone marrow.1 The most important growth factors for eosinophils are interleukin 3 (IL-3), IL-5 and granulocyte-macrophage colony-stimulating factor. They not only trigger growth but also activate normal and neoplastic eosinophils.1,4-5 There are two pathophysiological mechanisms that can contribute to the development of HE. First, overproduction of eosinophilopoietic cytokines can cause an increase in differentiation and massive proliferation. Subsequently, there is an increase in migration, adhesion, activation and survival of the eosinophils. 1,4,6 The second mechanism includes rapid monoclonal proliferation of eosinophils from myeloid progenitor cells caused by gene rearrangements of oncogenic tyrosine kinase receptors, such as PDGFRA, PDGFRB or FGFR1 (table 2).1,3,6 Fusion of the PCM1-JAK2 gene was added as a provisional entity in 2016.3 These mutated receptors induce constitutive tyrosine kinase activation, resulting in uncontrolled stimulation of eosinophilic progenitor cells.1 CEL is often caused by deletion of 4q12, resulting in the FIP1L1/PDGFRA fusion gene. Reactive hypereosinophilia can thus develop due to overproduction of cytokines and growth factors, or be the result of clonal eosinophilia caused by myeloproliferative disorders.
In the setting of persistent and massive activation, eosinophils can invade target organs and release their toxic mediators.1,5 These eosinophil-derived substances may cause profound changes in the microenvironment and can lead to resultant endomyocardial fibrosis, thrombosis, cutaneous symptoms, peripheral or central neuropathy (with chronic of recurrent neurologic deficit) and other less common organ manifestations.1
Etiology
Many conditions are associ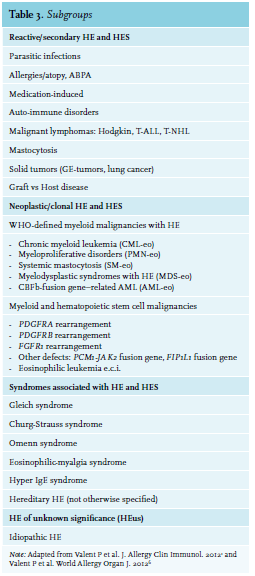 ated with HE and HES, and can be divided into several subgroups in which reactive HE and HES occurs most frequently (table 3).6 Worldwide, parasitic infections are the major cause of HE (table 4), with the most prevalent being hookworms, ascariasis and filariasis infections, followed by schistosomiasis and strongyloidiasis.7 In areas where there is little exposure to parasites, allergies and medication-induced HE are the leading cause.8,9 Solid tumors and autoimmune disorders occur less frequently. Finally, only a small proportion is caused by the remaining other syndromes shown in table 3;5,6 unfortunately, the exact relation between these syndromes and the development of HE is often obscure.
ated with HE and HES, and can be divided into several subgroups in which reactive HE and HES occurs most frequently (table 3).6 Worldwide, parasitic infections are the major cause of HE (table 4), with the most prevalent being hookworms, ascariasis and filariasis infections, followed by schistosomiasis and strongyloidiasis.7 In areas where there is little exposure to parasites, allergies and medication-induced HE are the leading cause.8,9 Solid tumors and autoimmune disorders occur less frequently. Finally, only a small proportion is caused by the remaining other syndromes shown in table 3;5,6 unfortunately, the exact relation between these syndromes and the development of HE is often obscure.
If no obvious ‘reactive’ mechanism is identified, further exploration for a possible hematological malignancy is indicated. Malignancies associated with hypereosinophilia include chronic eosinophilic leukaemia, myeloproliferative disorders (chronic myeloid leukaemia or polycythaemia vera), variants of acute myeloid leukaemia (AML) and some lymphoproliferative disorders (Hodgkin lymphoma and T-cell lymphomas) and systemic mastocytosis.3,6 In the case of myeloid disorders, eosinophilia is clonal and part of the myeloid malignancy. On the other hand, when there is an underlying lymphoproliferative disorder, eosinophilia is polyclonal and frequently due to overproduction of IL-5. Sometimes, the underlying pathology of HE remains unclear even after extensive examination. In this case, the term ‘hypereosinophilia of unknown significance’ (HEUS) is used.1 The main characteristic of HEUS is the absence of eosinophil-related organ damage. 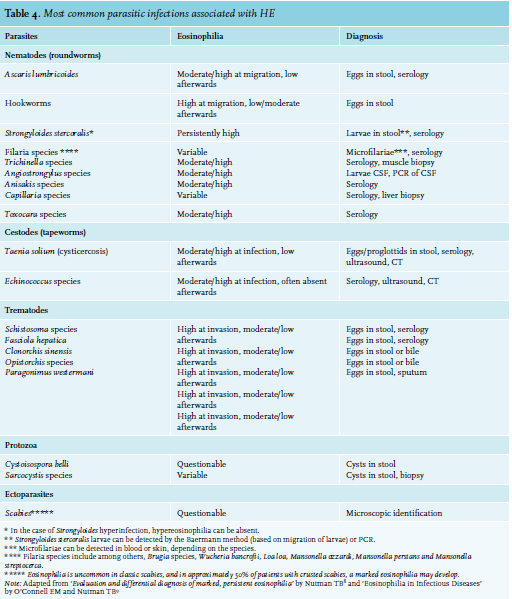 Clinical manifestations
Clinical manifestations
Depending on the underlying mechanism of HE or HES, a variety of clinical symptoms can occur. Essentially all organ systems can be affected, primarily skin, lungs, the digestive tract and heart.6 Most common presenting symptoms are fatigue (26%), coughing (24%), dyspnea (16%), myalgias and angioedema (14%), rash or fever (12%) and rhinitis (10%).3 Neurological deficits are also described, including muscle weakness and polyneuropathy. Special care should focus on the development of endomyocardial fibrosis (which is associated with risk of progressive heart failure) and thrombosis.
Diagnosis
Figure 1 presents an algorithm for the diagnostic work-up of HE which can be used in any clinical setting. Atopic constitution is often obvious from history and physical signs. An elevated serum level IgE or more specific tests such as radioallergosorbent or epicutaneous testing, further support this diagnosis. Pulmonary eosinophilia, which will not be discussed here, is a rare entity with both non-infectious (like allergic bronchopulmonary aspergillosis, acute and chronic eosinophilic pneumonias, Churg-Strauss syndrome) and infectious etiologies (mostly parasitic).3 A detailed history of used medication is important and should include the use of over-thecounter drugs, vitamins and supplements. For instance, in America, an epidemic of Eosinophilia–myalgia syndrome was linked to the use of L-tryptophan consumption.10
A travel history with sufficient exposure warrants a search for parasites. Serological methods are more sensitive and practical than the demonstration of eggs/cysts in stool. Particularly, HE is the immunological response to circulating worm antigens, which is most profound during the initial phase of infestation when larvae migrate through the organs and tissues of the host.7-9 Production of eggs detectable in varying quantities in stool samples is often apparent weeks later. When suspicion remains high, repeated testing is indicated.
Bone marrow examination is indicated in all patients with unexplained and persistent HE and should include cytological assessment of the aspirate, immunohistochemistry, cytogenetics, FISH and molecular analysis. Screening of rearrangements of PDGFRA, PDGFRB and FGFR1 is essential.3 The identification of the FIP1L1-PDGFRA fusion gene has to be performed with FISH or reverse transcription polymerase chain reaction (RT-PCR).
Furthermore, it is clinically relevant for all patients with persistent HE to screen for progression to HES and secondary organ failure. This often demands a multidisciplinary approach with different specialists in a consultative role. A thorough work-up by a cardiologist (echocardiography), dermatologist (skin biopsy) and neurologist may be advised. 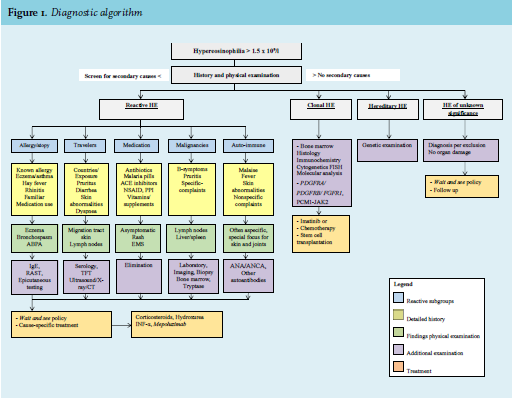 Treatment and follow up
Treatment and follow up
The first step in the management of HE is to identify and stop exposure to potential triggers such as medication and allergens. Parasitic infections require specific treatment, depending on type and stage of infection. Adequate treatment of Strongyloides infection with ivermectin is important because of the risk of Strongyloides hyperinfection.7 Repeated courses may be necessary to achieve full eradication. Empiric treatment with ivermectine 0.22 mg/kg in case of urgent need to initiate corticosteroid therapy, while results of serology are still pending, is justifiable. For patients with mild HE without signs of secondary organ damage, a wait-and-see policy can be justified.
Regular follow-up is essential to evaluate progression to HES with subsequent organ damage. If treatment is indicated, systemic corticosteroids (0.5-1 mg/kg) are highly effective in rapidly reducing the eosinophil count.3,6 Concomitant use of corticosteroid-sparing agents, such as hydroxurea and interferon-α, is often necessary because of side-effects and/or intolerance.5 Mepolizumab, an anti-IL-5 antibody, appeared to be safe and effective but is not currently approved by the Food and Drug Administration for HE(S).3,6,11 However, exceptional use for individuals with life-threatening HES who failed prior therapies, is described.
Patients with underlying hematological malignancies will generally fail to respond to steroids. Detailed discussion of different therapies is out of scope here. Referral to, or consultation of, a center of expertise is then recommended. When a PDGFRA or PDGFRB fusion gene is established, treatment with imatinib can be started (tyrosine kinase inhibitor).3,11 A low dose of 100 mg per day is often sufficient to achieve molecular remission. Prompt treatment is necessary to prevent irreversible complications such as endomyocardial fibrosis and thrombosis.6 Patients with a FGRF1 mutation are often resistant to treatment with imatinib (response varies between 14-60%). Intensive chemotherapy followed by allogeneic stem cell transplantation is often recommended.3
CONCLUSION
Hypereosinophilia is a rather frequently identified as laboratory abnormality. In most cases, there is an obvious identifiable cause, but sometimes more extensive exploration is necessary. A detailed history is still the most important first step in diagnosis. We must be aware of hematological malignancies and not hesitate to perform a bone marrow examination when the diagnosis remains unclear. Finally, in the case of persisting HE, we must always be aware of the progression to HES.
REFERENCES