KEYWORDS
Neuroendocrine tumour, carcinoid, peptide receptor radionuclide therapy
INTRODUCTION
Despite their reputation as uncommon malignancies, neuroendocrine tumours (NETs) are nowadays increasingly encountered across the practices of endocrinologists, oncologists, gastroenterologists, surgeons and pulmonologists.1 Different tumour subtypes at multiple sites are linked due to their common origin of neuroendocrine cells within the pulmonary and gastrointestinal tracts. Research has clearly delineated the separate genetic backgrounds and biological behaviours of NETs originating at different primary locations in terms of proliferative potential and hormone production. These insights have resulted in a paradigm shift in patient treatment according to tumour subtype and clinical syndrome. Consequently, any medical professional dealing with NETs should be well informed about the NET phenotypes and how the emerging therapies can be best applied to the individual patient suffering from this disease.
EPIDEMIOLOGY
NET is a rare malignancy that comprises 1-2% of all gastrointestinal and pulmonary malignancies.2,3 A prospective, nationwide study on the occurrence of gastrointestinal and pancreatic NETs in Austria revealed an annual incidence ratio of 2.4 per 100,000 persons.2 According to a national cancer registry in the UK the incidence of gastrointestinal NETs is 1.3 per 100,000 and has markedly risen during the past four decades.4 Recently, a large population-based USA registry showed an incidence of all NETs of 7.0 per 100,000 persons in 2012, which is over 6-fold higher compared with the 1970s. This increment was seen across all age groups, tumour sites, stages and grades. In the past two decades, the 20-year limited-duration prevalence of NETs also rose 8-fold to 0.048%.5 The increased detection of the disease is likely explained by the rise in the use of endoscopic investigations and cross-sectional imaging combined with an increased awareness among pathologists to consider a diagnosis of NET.
TUMOUR SITES, GRADING AND STAGING
Classically, NETs have been subdivided according to the origin of the primary tumour.6 Tumours were historically classified as arising from the embryonic foregut, midgut or hindgut. Foregut NETs comprise lung, thymic, stomach, duodenal and pancreatic tumours. Midgut NETs arise from the neuroendocrine cells of the small intestine distal to the duodenojejunal flexure, the appendix and the ascending colon. NETs of the hindgut originate in the distal colon or rectum. Tumours originating from the gastrointestinal tract are commonly called gastroenteropancreatic NETs (GEP-NETs). Despite contemporary imaging techniques, about 5-10% of metastasised NETs have an occult primary tumour.7 Neuroendocrine neoplasms can arise in organs outside the pulmonary and digestive tract and features of neuroendocrine differentiation can be observed in a subset of common malignancies, such as breast and prostate cancer, but these neoplasms are generally not classified as NET.
One of the key features of NET is the considerable difference in biological behaviour ranging from indolent rectal tumours to the highly malignant small cell lung cancer. As such, grading of NETs is crucial for estimation of prognosis and for choice of appropriate anti-proliferative management. The current World Health Organisation (WHO) grading system divides NETs into three subgroups depending on histopathological evaluation of the mitotic index and the Ki67 index.8,9 In the case of lung NETs the presence of necrosis is also taken into account.10 Tumours with grade 1 (low) or grade 2 (intermediate) are generally considered well-differentiated and are accompanied by the best prognosis. Grade 3 or poorly differentiated tumours with a high mitotic and/or Ki67 index display aggressive behaviour, which is why this subtype is also classified as neuroendocrine carcinoma (NEC). An additional tumour group of grade 3 NET is incorporated in the new WHO 2017 grading system which is comprised of histologically well-differentiated tumours that harbour a high proliferative index, i.e. Ki67 index above 20%. This review will focus primarily on well-differentiated tumours. Staging occurs according to a TNM-based system with localised disease representing stages I and II, invasion into adjacent structures (T4) or lymph node metastases (N1) denoting stage III and distant metastases (M1) are the hallmark of stage IV disease.8,9
PATHOPHYSIOLOGY
In a minority of cases, NETs can develop within the context of several inherited syndromes.11 Notably, patients with multiple endocrine neoplasia type 1 (MEN1), caused by germline mutations in the MEN1 gene, have a predisposition for developing pancreatic and bronchopulmonary NETs. Hereditary susceptibility to pancreatic NETs (PanNETS) is also encountered in Von Hippel-Lindau disease, tuberous sclerosis complex and neurofibromatosis type 1.
Similar to other malignancies, much has become known about disease-causing genes through next-generation sequencing studies. First, exome sequencing of PanNETs revealed a predominance of mutations in MEN1 (44%) as well as mutations in the interacting telomere-altering genes ATRX (α thalassaemia/mental retardation syndrome X-linked) and DAXX1 (death-domain-associated protein) in a mutually exclusive pattern in 43% of patients.12 Signalling of the kinase mammalian target of rapamycin (mTOR) is involved in a subset of patients, as pathway-activating mutations have been detected in 15% of PanNETs. Recently, whole-genome sequencing of PanNETs identified four mutational signatures revealing pathogenic alterations in pathways of chromatin remodelling, DNA damage repair, the mTOR pathway and telomere maintenance.13
For midgut or small intestinal NETs exome sequencing identified a variety of single nucleotide mutations with low penetrance; gene copy number alterations appeared to be more common.14 Genetic changes were clustered within several growth factor pathways, such as the transforming growth factor-β (TGF-β), platelet-derived growth factor and epidermal growth factor. Inactivating alterations in the cell cycle regulator CDKN1B (cyclin-dependent kinase inhibitor 1B), the gene encoding p27 and responsible for MEN type 4, were the most prevalent at 8% of tumours.15 Similar to PanNETs, signatures of genetic aberrations detected in midgut NETs were grouped in the mTOR pathway, DNA damage repair and chromatin remodelling.14 Further studies have confirmed that small intestinal NETs appear to be caused by epigenetic rather than genetic dysregulation.16
In well-differentiated lung neuroendocrine tumours, also termed bronchial carcinoids, there is again a molecular signature involving chromatin-remodelling genes.17 Prevalence of disease-causing mutations therein was high with over 60% of tumours affected and was accompanied by significant loss of heterozygosity at several affected genes. Consequently, alterations in chromatin-remodelling, mTOR pathway and DNA repair mechanisms appear key factors in the pathogenesis of NETs.
HORMONAL SYNDROMES
NETs can be divided into ‘functional’ and ‘non-functional’ tumours. Functional NETs produce bioactive peptides and hormones resulting in specific symptoms whereas non-functional NETs can present with mechanical effects, i.e. bowel obstruction or ischaemia. Non-functional NETs are also frequently discovered incidentally during diagnostic procedures. Patients with functional NETs can present with a range of clinical symptoms related to hormonal secretion by the tumour. Next to production of hormones that are physiologically produced by neuroendocrine cells, functional NETs can also produce hormones that are normally secreted by endocrine glands, i.e. ectopic hormone production. Patients should be asked about the occurrence of these symptoms as they require specific treatment and can potentially impair prognosis.18
The classic hormonal syndrome encountered in patients with NETs is the carcinoid syndrome.19 Patients present with cutaneous flushes and diarrhoea due to vasodilation and increased gut motility, respectively. NETs can secrete a variety of amines and peptides that elicit these symptoms, but serotonin or 5-hydroxytryptamine is the most well-known mediator causing carcinoid syndrome.20 It is produced by the enterochromaffin cells in the small intestine and its overproduction is predominantly encountered in midgut NETs, although primary tumours at other sites can also secrete this amine. Besides its vasodilatory and peristaltic effects, serotonin is also the most likely mediator producing mesenteric and right-sided cardiac fibrosis, which are exclusive features of carcinoid syndrome.21
PanNETs arise from the islets of Langerhans and as such are able to produce pancreatic peptides. Hypoglycaemia can be a severe and potentially life-threatening complication of insulin-producing PanNETs also known as insulinomas.22 Physicians should also be aware of Zollinger-Ellison syndrome, caused by a gastrin-producing duodenal or pancreatic NET with clinical sequelae of severe gastric acid hypersecretion, i.e. multiple peptic ventricular or duodenal ulcers.23 Glucagon release by the tumour can lead to diabetes mellitus, cachexia, glossitis and a typical rash termed necrolytic migratory erythema.24 Production of vasoactive intestinal peptide (VIP) or calcitonin by PanNETs can trigger severe life-threatening secretory diarrhoea.25 Rare clinical syndromes encountered in NETs are caused by ectopic hormone production including hypercalcaemia due to parathyroid hormone-related peptide (PTHrP),26 the syndrome of inappropriate antidiuretic hormone, Cushing’s syndrome due to adrenocorticotrophic hormone27 and acromegaly caused by growth hormone-releasing hormone.28
DIAGNOSIS
A diagnosis of NET can be suspected because of incidental findings during endoscopic or cross-sectional imaging or following symptom-directed investigations into the sequelae of tumour mass or hormone release. Central to the diagnosis of all NETs is histological evaluation of tumour tissue.29 Primary or metastatic lesions should be biopsied or resected when feasible for confirmation of diagnosis and determination of the tumour grade. Histologically, well-differentiated NETs display an organoid arrangement of cells, with a nesting, trabecular or gyriform pattern and stain positive for neuroendocrine markers, particularly synaptophysin and chromogranin A.6 These immunohistochemical markers are key to diagnosis and their increased application has likely contributed to the observed rise in incidence of NET.5 In the case of an unknown primary tumour, staining for TTF1 (lung NET), CDX2 (midgut NET) and ISL-1 or PDX-1 (PanNET) can guide the pathologist and clinician towards a likely source.29
When the diagnosis is confirmed, routine measurements of glucose, renal and liver function, calcium and blood cell counts should be accompanied by determination of chromogranin A (CgA) levels and, in case of highly proliferative NET, neuron-specific enolase levels. Sensitivity of serum CgA in NET patients is reported to be 50-90% with an average of 73%,30-32 but for those patients with elevated levels these markers can be used for follow-up as they may represent changes in tumour volume or biology. Importantly, false-positive findings are frequently encountered, since elevated levels can be caused by medication, most notably proton pump inhibitors, atrophic gastritis or renal failure.33 As such, clinicians should refrain from CgA measurements for NET screening in patients with a low a priori probability of NET. Hormonal analysis should be guided by symptoms; in the case of an apparent clinical syndrome dedicated investigations are indicated with measurement of the serotonin metabolite 5-hydroxyindolacetid acid in 24-hour urine collection, insulin and C-peptide during hypoglycaemia in a supervised 72-hour fast, glucagon, gastrin, VIP, PTHrP, 24-hour urinary free cortisol excretion, cortisol after 1 mg dexamethasone overnight or insulin-like growth factor 1. If the family history is suspicious or positive for hereditary NET-related syndromes, dedicated genetic testing should be requested accordingly.
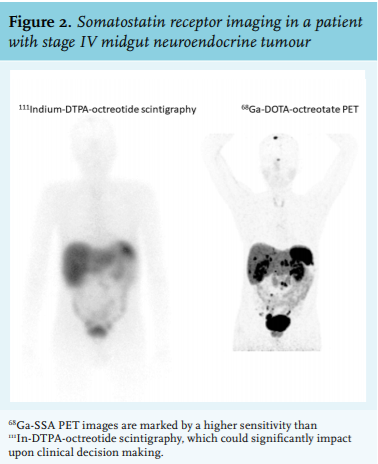
Tumour staging for the presence of metastases should be performed in all NETs (figure 1). Low-grade gastric or rectal NETs smaller than 1 cm carry an excellent prognosis and for these tumours staging with endoscopic ultrasound will be sufficient.34,35 In all other cases, cross-sectional imaging with computer tomography (CT) or magnetic resonance imaging (MRI) is preferred as initial staging.34-39 On top of this, somatostatin receptor imaging is advised. Low-grade and a subset of high-grade NETs have a high expression of the somatostatin (SS) type 2 receptor (SSTR2A) which can be used for molecular imaging. Following the introduction of somatostatin receptor scintigraphy (SRS) with 111In-DTPA-octreotide (Octreoscan®) in the 1980s, this modality has been extensively used due to its superiority over cross-sectional images for NET.40 Besides tumour staging, the uptake on scintigraphy can assist in the management of patients, for example for the decision on local or systemic treatment. More recently, dedicated NET centres have adopted the use of 68Ga-DOTA-SS analogue positron emission tomography CT (PET-CT), which is more sensitive and patient-friendly than Octreoscan41 (figure 2). Implementing this diagnostic modality instead of anatomic imaging or Octreoscan® can change management decisions in up to a third of patients.7 SRS gradually becomes negative in more aggressive tumours such as NECs, limiting their yield in poorly differentiated tumours. Alternatively, glucose metabolism increases in these tumours, making them avid on 18fluoro-deoxyglucose PET imaging.42 As such, selective PET imaging can be applied according to the tumour grade, whereas some centres even advise to perform imaging with both PET tracers. Additionally, radio-labelled exendin-4, a glucagon-related peptide 1 receptor agonist, has specifically been developed for the detection of insulinomas. 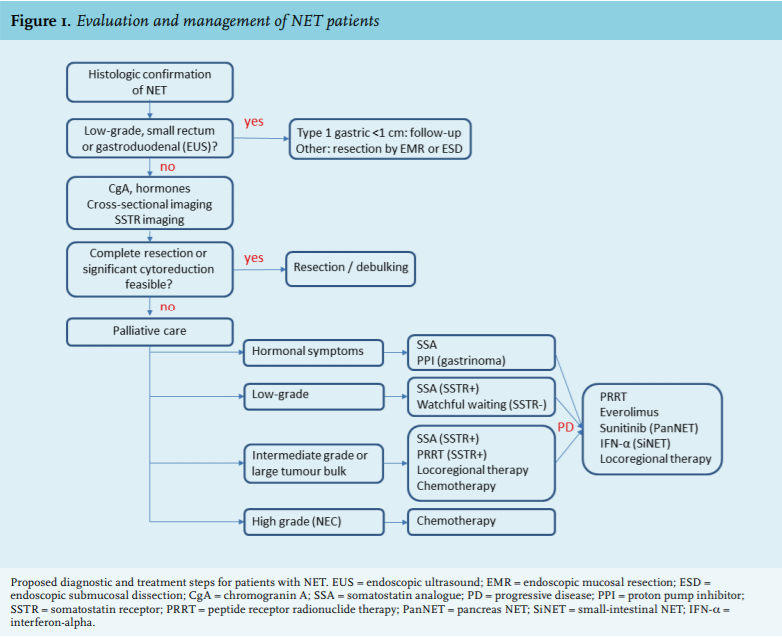
MANAGEMENT
The primary goal of the management of a patient with a NET is cure and consequently the possibility of complete resection should be evaluated.43,44 For small gastroduodenal NETs or colorectal NETs this might be achieved through endoscopic resection. As an exception, type 1 or atrophic gastritis-related gastric NETs below 1 cm can be managed conservatively with follow-up endoscopy.34 As many gastroduodenal and colorectal NETs grow into the submucosa, endoscopic mucosal resection or endoscopic submucosal dissection should be employed by an experienced gastroenterologist.34,35 Many NETs unfortunately present at advanced stages marked by unresectable or disseminated disease, with a predominance of liver and bone metastases.45 In selected cases of hepatic metastases, especially in young patients, complete surgical resection or ablation can still be attempted as surgical cytoreduction of NET carries an excellent prognosis.46 If surgical resection is not considered feasible, tailored palliative care should ensue. Again, tumour grading is highly relevant as some grade 1 tumours might not show growth over time whereas most grade 3 tumours require aggressive anti-proliferative treatment.
Treatment of well-differentiated NETs with conventional chemotherapy has little success. For most grade 3 NECs chemotherapy with platinum-based regimens with etoposide is the first-line treatment of choice.47 Regimens including the DNA-alkylating agents streptozocin or temozolomide have been studied and can be considered for progressive and/or bulky and/or symptomatic intermediate- to high-grade PanNETs and NECs.48,49 Temozolomide with or without capecitabine has also been studied in single arm or retrospective series of well-differentiated non-pancreatic NETs, where encouraging responses have been observed.50 The indolent nature in the majority of well-differentiated NETs clearly make them unsuitable for toxic chemotherapy and targeted treatment has been highly desired. Only in the last decade have significant improvements been made in this field based on large, international, randomised clinical trials with hormonal or targeted therapy (figure 3).
As stated before, NETs are generally characterised by a high expression of SSTR, although expression levels may vary considerably among tumours and between different types of tumours. To date, five SSTR subtypes have been identified.51 The SSTR2A subtype is expressed at the highest level and explains the successful application of the somatostatin analogues (SSA) octreotide and lanreotide, which are analogues preferentially binding to SST2, in the medical treatment of patients with NET. Specific treatment of hormonal symptoms started in the 1980s with octreotide, an SSA with a favourable half-life compared with its natural counterpart. Subcutaneous injection of octreotide decreased carcinoid syndrome-related symptoms in the majority of patients.52 Nowadays, long-acting SSA octreotide and lanreotide are indicated for all patients with carcinoid syndrome in the palliative setting and can be implemented for other NET-related hormonal syndromes.45 SSTR imaging should be employed to check for tumoral SSTR2A status. Importantly, patients with carcinoid syndrome who undergo surgery should be treated with high-dose intravenous SSA in order to prevent a carcinoid crisis, a state of haemodynamic instability due to the massive release of vasoactive hormones.
At the time of SSA introduction, it was also found that daily interferon-alpha (IFN-α) injections ameliorated hormonal symptoms and produced modest anti-proliferative effects in midgut NETs. IFN-α treatment, however, is accompanied by extensive side effects, most notably flu-like symptoms.53 For gastrinomas, treatment with proton pump inhibitors successfully diminished acid-related complications and has become the first-line treatment of choice.23 As somatostatin analogues do not abolish clinical symptoms in all patients with carcinoid syndrome, additional treatment is required beyond toxic cytoreductive strategies. A recently developed drug that inhibits serotonin synthesis, telotristat ethyl, offers novel options as it decreased carcinoid syndrome-associated diarrhoea and flushes in a subset of patients.54
Apart from suppression of hormone secretion, clinical observations have shown that SSA may also inhibit tumour growth in patients with NET. This led to the first placebocontrolled, double-blind clinical trial in NET. In the PROMID trial octreotide LAR was compared with placebo in SSTR-positive midgut NET patients; 95% of the tumours were grade 1.55 Patients receiving the SSA had a median progressionfree survival of 14 months as compared with 6 months in the placebo group, whereas overall survival was not affected. This was followed by the CLARINET trial in which patients with SRS-positive enteropancreatic NETs and a Ki67 below 10% were randomised to treatment with long-acting lanreotide or placebo.56 Lanreotide also significantly delayed progressionfree survival to a median of 33 months compared with 18 months in patients receiving placebo.57
The use of gamma-emitting radiolabelled SSAs in NET diagnostics paved the way for treatment with beta-emitting radiolabelled SSAs. Peptide receptor radionuclide therapy (PRRT) has mainly been performed using the radionuclides 90Yttrium-DOTA-octreotide and 177LutetiumDOTA-octreotate. The latter beta-emitting radionuclide has a favourable safety profile with regard to renal and haematological toxicity and has been applied by most dedicated centres in the last 15 years. After many retrospective series, the first randomised, multicentre phase 3 trial was recently published.58 The NETTER-1 trial proved the efficacy of PRRT with 177LutetiumDOTA-octreotate in patients with well-differentiated, SSTR-positive midgut NETs that were progressive on a standard dose of octreotide LAR. Compared with patients on high-dose octreotide LAR patients receiving PRRT had an increase in progression-free survival. Although an objective response was only obtained in 18% of patients treated with PRRT, it predominantly induces prolonged tumour stabilisation. PRRT with 177Lutetium-DOTA-octreotate was well tolerated with mostly transient side effects. Similar survival data have been shown for SSTR-positive NETs of other origins, with the best responses obtained in PanNETs.59 Besides the anti-proliferative outcomes, PRRT positively affected quality of life, symptoms and functioning in patients.60 Concerns for long-term sequelae have been raised with reports on increased occurrences of myelodysplastic syndrome and acute leukaemia, but the latest data show that these risks for 177Lutetium-DOTA-octreotate are acceptable at 1.5% and 0.7%, respectively, after a median follow-up time of 64 months.61 No therapy-related renal failure was observed.
In recent years, we have also seen the advent of oral molecular targeted therapy. As NETs are typically hypervascularized, treatment with anti-angiogenic drugs has also been evaluated for NET. The tyrosine kinase inhibitor sunitinib, which displays activity against VEGF receptors amongst others, has been shown to increase progression-free and overall survival in patients with advanced, well-differentiated PanNET,62 making this an option for patients with progressive disease. Sunitinib appeared less effective in patients with advanced lung or midgut NETs.63 The key role of the mTOR pathway in the pathogenesis of NET has fuelled trials with everolimus, an mTOR inhibitor. Growth stabilisation by everolimus has been demonstrated in both pancreatic NETs64 as well as in lung and gastrointestinal NETs.65 In two pivotal trials in patients with advanced, well-differentiated, progressive tumours everolimus increased progression-free survival to a median of 11 months compared with four or five months in patients taking placebo.
Given the indolent nature of NETs locoregional therapy, particularly targeting the liver, can be administered. Due to the predominant vascular supply by the hepatic artery, NETs are particularly susceptible for treatment with embolisation procedures. Options for treatment of NET metastases include radiofrequency ablation, transarterial embolisation or transarterial chemoembolisation.66 All three can provide tumour relief and improvement of hormonal symptoms in patients; no large series have compared efficacy between these treatments in NETs. A recent development incorporates radiolabelling of microspheres with 90Yttrium or 166Holmium for local radioembolisation of liver metastases, a technique known as selective internal radiation therapy.67
Choices among these different treatment options should preferably be discussed in a multidisciplinary team meeting with participants who have ample experience in dealing with NET (figure 1). Advanced non-functioning, asymptomatic, low-grade tumours can be safely monitored with a watchful waiting policy employing longitudinal imaging as a subset of these tumours shows little if any spontaneous growth. In low to intermediate grade or functional NETs that are SSTR positive, treatment with a long-acting SSA is generally preferred due to its tumour-stabilising and anti-hormonal effects accompanied by a good tolerability. In case of tumour progression on SSA therapy the optimal treatment sequence is unclear due to the lack of data. However, the progression-free survival and responses obtained with PRRT appear to exceed that of targeted therapy and at no significant increase of toxicity, making this a reasonable second-line option. Following progression after PRRT or in SSTR-negative tumours, everolimus or in the case of PanNET sunitinib can be administered. Patient characteristics and the ever-changing behaviour of individual tumour lesions should always be taken into account, necessitating a comprehensive view in an experienced multidisciplinary team.
PROGNOSIS
For many years, the lack of specific treatment options for NETs was instrumental in a disappointing outcome and prognosis for patients, despite the indolent nature in a subset of tumours. Consequently, median overall survival for all NETs between 1973 and 2004 was only 75 months.68 By 2012, these estimates had already increased to up to 112 months, with most notable improvements detected in patients with advanced GEP-NET.5 Presumably, the availability of improved diagnostic and therapeutic strategies together with the centralisation of care into dedicated NET centres have contributed to better patient outcome. Concerning the latter, several specialised NET centres have published their data on overall survival for stage IV, well-differentiated GEP-NETs which exceeds 100 months.69,70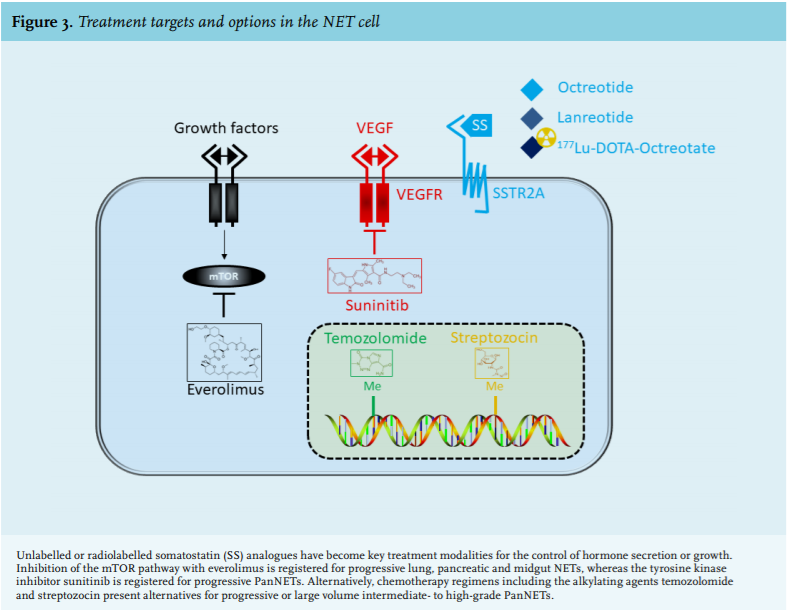
CONCLUSION
Several milestones have emerged recently in the NET field with the improvement in understanding of the genetic background, superior diagnostic modalities and the first randomised, multicentre clinical trials. This provides the clinician with emerging options for their patients with a NET, which can improve both survival and hormonal symptoms. We are only at the brink of properly understanding the heterogeneity of this disease and how to predict which patients will respond to particular therapies. Hopefully, further insights into tumour biology, including the epigenetics and control of hormonal stimuli, will pave the way towards optimal patient treatment strategies for NETs in the future.
DISCLOSURES
Grant support: J. Hofland is supported by the Daniel den Hoed foundation.
REFERENCES