KEYWORDS
Everolimus, kidney, randomised-controlled trial, tacrolimus, transplantation
INTRODUCTION
Everolimus is an immunosuppressive drug that lacks the chronic nephrotoxic effects of the calcineurin inhibitors (CNIs) tacrolimus and cyclosporine and has the potential to improve long-term outcomes of kidney transplantation.1,2 In the first clinical trials, everolimus was combined with cyclosporine in de novo kidney transplant recipients. These trials did not demonstrate a significant improvement in renal function compared with standard CNI-based immunosuppression. Everolimus was found to have considerable toxicity, including delayed wound healing, the formation of lymphoceles, dyslipidaemia and cytopenia.2-4 An alternative strategy that has been tested in clinical studies is to convert patients from a CNI-based immunosuppressive regimen to an everolimus-based immunosuppressive regimen longer (i.e. >1 year) after transplantation. The results of these studies have been disappointing as the majority failed to demonstrate a significant improvement in renal function.5,6
At present, switching kidney transplant recipients sometime after the critical early post-transplant phase, when rejection risk is highest, from a CNI to everolimus seems to have the most potential in terms of improving long-term renal transplant function without risking excess acute rejection. In the randomised, controlled ZEUS trial, patients were randomised to either continue cyclosporine or were converted to everolimus 4.5 months after transplantation.7 This trial demonstrated that conversion to everolimus resulted in superior renal function 1, 3 and 5 years after transplantation, despite a moderately increased risk of acute rejection (13.6% vs. 7.5% after 5 years).7-9 However, in the ZEUS study, everolimus was compared with a cyclosporine-based immunosuppressive regimen.7 At present, in most transplant centres in the United States and in Europe, tacrolimus is the cornerstone immunosuppressant. It remains to be determined if switching from tacrolimus to everolimus will result in equally good outcomes and what the optimal timing of such a conversion would be.
The objective of this randomised, controlled clinical trial was to investigate if conversion from a tacrolimusbased immunosuppressive regimen to an everolimusbased regimen at month 3 after living-donor kidney transplantation in low to moderate immunological risk patients with complete and early elimination of glucocorticoids results in an improvement of renal transplant function.
MATERIALS AND METHODS
Study design
This was an investigator-initiated, prospective, randomised, controlled, parallel group, open label, single centre trial that was conducted in the Erasmus MC, University Medical Centre Rotterdam, the Netherlands.
Adult patients (≥ 18 years) who received a blood group AB0-compatible kidney transplant from a living donor (excluding HLA-identical siblings), who were transplanted in our hospital and on continued follow-up in our clinic, were eligible for participation. The patients had to be treated with immunosuppressive therapy consisting of tacrolimus, mycophenolate mofetil and prednisolone at month 3 after transplantation. All patients received induction therapy with basiliximab (Simulect®, Novartis Pharma B.V., Arnhem, the Netherlands) in a dose of 20 mg intravenously on days 0 and 4. None of the patients received induction therapy with lymphocyte depleting antibodies.
Exclusion criteria were 1) an acute rejection episode less than 4 weeks prior to the planned randomisation; 2) proteinuria ≥ 1.0 g/day; 3) estimated GFR (eGFR) ≤ 30 ml/min; 4) recipient of multiple organ transplants; 5) a positive pre-transplant complement-dependent cytotoxicity cross-match; 6) human immunodeficiency virus seropositivity; 7) recipients of an allograft from a hepatitis B surface antigen or a hepatitis C virus seropositive donor; 8) severe allergy / hypersensitivity to drugs similar to everolimus (such as macrolides); 9) severe, uncontrollable hypercholesterolaemia or hypertriglyceridaemia; 10) a white blood cell count ≤ 2000/mm3 or a platelet count ≤ 50,000/mm3 ; 11) ongoing wound healing problems; 12) clinically significant infections; 13) severe surgical problems in the opinion of the investigator; 14) intractable immunosuppressant complications or side effects; 15) pregnant or lactating patients; 16) patients who were planning to become pregnant or were unwilling to use effective means of contraception. Donor-specific anti-HLA antibodies were not measured at the time of inclusion (nor thereafter during the course of the trial) and were thus not considered as a possible exclusion criterion.
Interim analyses were planned after the inclusion of 60 patients and again after the inclusion of 120 patients. A data safety monitoring board was instituted to analyse the interim analyses and decide on continuation or modification of the trial.
The study was approved by the institutional review board of the Erasmus MC (Medical Ethical Review Board number 2010-235) and was registered in the Dutch National Trial Registry (http://www.trialregister.nl/trialreg/index.asp; number: NTR2545, registered 6 September 2010). Written informed consent was obtained from all patients before randomisation. The study was performed in compliance with the Good Clinical Practice guidelines and in accordance with the declaration of Helsinki.
Intervention and randomisation
The patients were enrolled and randomised on a 1:1 basis by one of the coordinating investigators (R.B., N.S., T.v.G., or D.A.H.) to either continue tacrolimus or to switch to everolimus-based maintenance immunosuppressive therapy. The randomisation was performed by use of sealed, opaque, sequentially-numbered envelopes containing treatment allocation. The random-allocation sequence was generated by an independent statistician using a random number generator on a computer. Data were collected, monitored and entered by the coordinating investigators and stored in a hospital-based electronic study database.
All patients received tacrolimus (Prograf®, Astellas Pharma, Leiden, the Netherlands), mycophenolate mofetil (Cellcept®, Roche Pharmaceuticals, Basel, Switzerland) and prednisolone triple immunosuppressive therapy at the time of enrolment and randomisation which was month 3 ± 3 weeks. After randomisation, patients either continued treatment with tacrolimus (aiming for pre-dose concentrations of 5-10 ng/ml) or were converted to everolimus (Certican®, Novartis Pharma B.V., Arnhem, the Netherlands) therapy. The everolimus starting dose was 1.5 mg twice daily and thereafter the everolimus dose was adjusted aiming for whole blood pre-dose concentrations of 4-7 ng/ml. Tacrolimus was reduced to 50% on the day of initiation of the everolimus therapy. One week after the introduction of everolimus, tacrolimus was withdrawn. Following our standard immunosuppressive protocol, prednisolone was tapered from 20 mg orally (started on day 3 after transplantation; all patients received 100 mg prednisolone intravenously for the first 3 days) to 5 mg over the course of the first three postoperative months. Prednisolone was tapered from 5 mg daily at the time of conversion to 0 mg in one month’s time following randomisation in both groups. The reason for complete glucocorticoid elimination in both arms was the fact that combination therapy of tacrolimus plus mycophenolate mofetil with complete cessation of glucocorticoids has been the standard of care in our centre for more than 10 years. Continuation of prednisolone in the control arm was therefore considered unethical.
Renal transplant biopsies
All patients included in this trial were asked to undergo a protocol biopsy at month 3 and again at month 12 after transplantation. However, this protocol biopsy was not mandatory and patients could be included in the trial without a baseline protocol biopsy. All biopsies (both for cause and protocol) were assessed locally by two pathologists (M.C.C.-v.G. and J.D.) and scored according to the most recent Banff criteria.10 For the trial reported here, only renal transplant biopsies to determine cause were considered and analysed.
Endpoints
The primary endpoint of the trial was renal function (eGFR) at month 12 ± 6 weeks after transplantation calculated by the 4-variable MDRD formula.11 Secondary endpoints were graft survival, the incidence of biopsy-proven acute rejection (BPAR) between month 3 and month 12 (based on for cause biopsy findings only), adverse events (AE), serious adverse events (SAE) and renal histology on protocol biopsy (including signs of CNI-related nephrotoxicity at month 12).
Safety
The incidence of adverse events was registered. An adverse event was defined as serious when 1) it necessitated or prolonged patient hospitalisation; 2) caused persistent or significant disability or incapacity; 3) was life-threatening; 4) caused the death of a patient or 5) required an intervention to prevent an event listed under point 1) to 4). Patients were followed until month 12 ± 6 weeks after transplantation.
Tacrolimus and everolimus concentration measurements
Tacrolimus concentrations were measured in ethylene diamine tetra-acetic acid (EDTA) blood using the affinity chrome-mediated flex-immunoassay (ACMIA) on a Dimension Xpand analyser (Siemens HealthCare Diagnostics Inc., Newark, DE) in accordance with the manufacturer’s instructions.12 Everolimus concentrations were determined using the sirolimus ACMIA kit from Siemens that highly cross-reacts with everolimus.13
Statistical analysis
It was estimated that a total of 194 patients had to be included in the trial in order to detect a difference in eGFR of 8 ml/min per 1.73 m2 between the two groups with a 90% power and accounting for a 30% dropout rate. Because the trial was terminated prematurely (after the first interim analysis), the focus of this report is on the safety aspects of conversion from tacrolimus to everolimus. Data on the primary endpoint (eGFR) will be presented for completeness.
For the analysis, an intention-to-treat approach was followed, which included all randomised patients who received at least one dose of the assigned drug. All summary statistics are presented by treatment group. Frequency distributions are provided for categorical variables. The two treatment groups were compared using X2 tests or Student’s t test to evaluate the null hypothesis of no difference in eGFR (and the secondary endpoints) between the tacrolimus and everolimus groups. For 2 x 2 tables, Yates’ correction for continuity was used. If the minimal expected value in a 2 x 2 table was below 5, Fisher’s exact test was used. The Shapiro-Wilk test was used to assess the normality of data. When this assumption was violated, the median and range are displayed and the Mann-Whitney U test was used to evaluate the null hypothesis of no relationship between secondary endpoints. All statistical tests were two-sided and used the 0.05 level of statistical significance. The statistical analyses were conducted using IBM SPSS Statistics version 21.0. Armonk, NY: IBM Corp.
Role of the funder
This was an investigator-initiated study. The trial was financially supported by Novartis Pharma B.V., Arnhem, the Netherlands, the producer of everolimus. Novartis Pharma B.V. had no role in the study design, data collection, data analyses, data interpretation, or writing of the report. All authors had full access to all the data, had final responsibility for the contents of this publication and the decision to submit for publication.
RESULTS 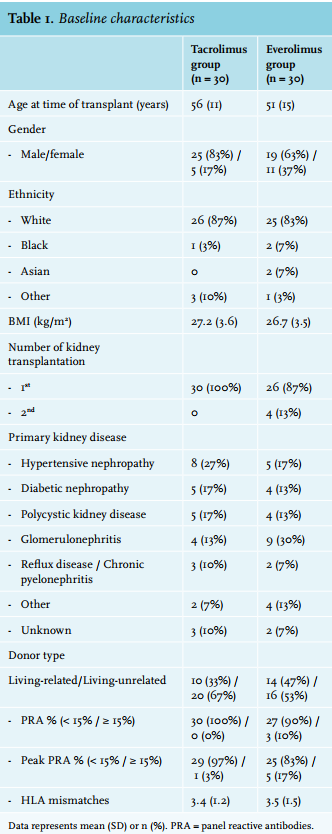
Patient population and trial progress
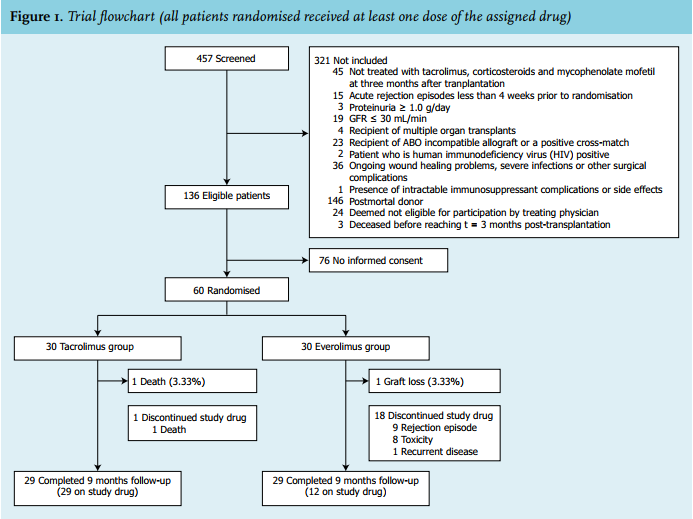
This study was conducted between 17 February 2011 (first patient, first visit) and 14 March 2014 (last patient, last visit). A total of 457 patients were screened for participation in the trial of which 136 were eligible, as shown in figure 1. Sixty patients gave written informed consent and were subsequently included and randomised. The characteristics of these 60 patients are summarised in table 1. Patients were enrolled in the study at a median of 96 days (range 83-111) after transplantation. A total of 58 patients (96.7%) completed the 9 month (± 6 weeks) follow-up. At the end of follow-up, 100% in the control group were on the assigned therapy, while only 40% of the patients in the intervention group were still on everolimus (p < 0.001). The primary reasons for discontinuing everolimus were acute rejection (number in group (n) = 9) and toxicity (n = 8); see figure 1 and below.
The trial was ended prematurely after the first, pre-planned interim analysis. The reasons for discontinuation were twofold. First, the interim analysis showed a significantly and unacceptably high incidence of BPAR in the everolimus group compared with the tacrolimus group: 30.0% vs. 6.7%; p = 0.042 (figure 2) (for details see below: under ‘Acute rejection’). Second, because the clinical condition of these patients required re-conversion to tacrolimus and because a considerable number of non-rejecting patients stopped taking everolimus for other reasons (see below), the overall dropout rate was 60%, which was much higher than the anticipated 30%. 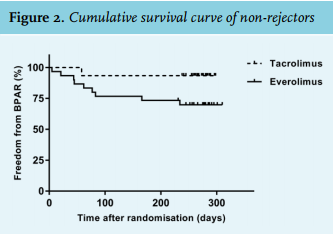
Acute rejection
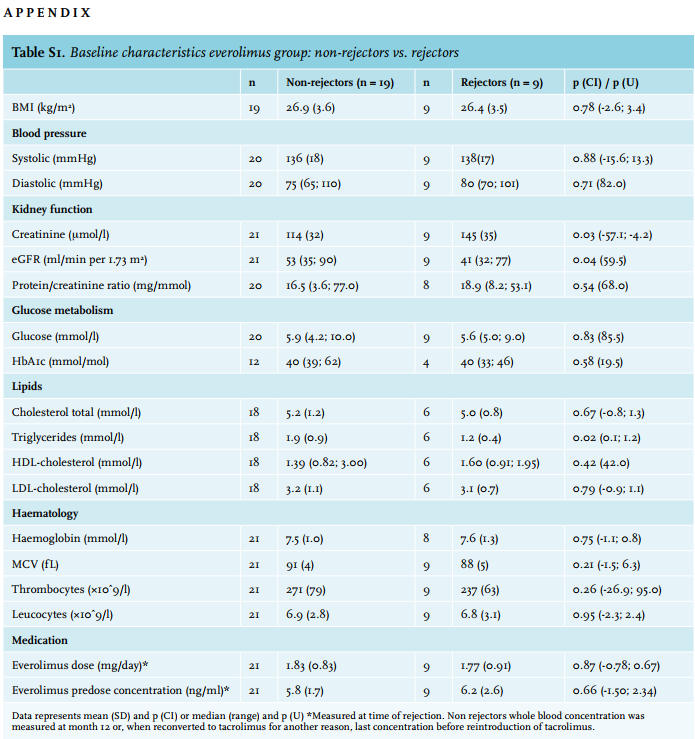
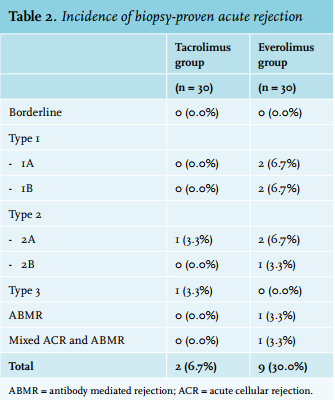
Overall, the BPAR rate in the everolimus group was 30.0% vs. 6.7% in the control group (95% CI: 0.048; 0.420; p = 0.045) (figure 2). Banff grades and frequencies are depicted in table 2. No cases of presumed acute rejection (i.e. clinically suspected rejection without histological confirmation) occurred. Baseline characteristics between rejectors and non-rejectors in the everolimus group are listed in Supplementary table 1. Estimated GFR at month 3 (baseline) was significantly lower among rejectors (U = 59.5; p = 0.04). There were no significant differences in everolimus dosages and concentration measurements between rejectors and non-rejectors.
Safety and tolerability
One patient (randomised to tacrolimus) died 272 days after transplantation due to metastasised gastric carcinoma.
This resulted in a patient survival of 97% and 100%, in the tacrolimus and everolimus groups, respectively (p = 0.41). One patient (randomised to everolimus) lost his graft as a result of uncontrollable acute rejection.
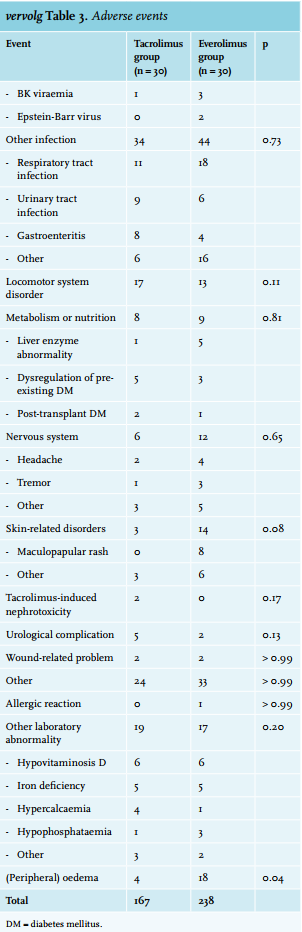
The AEs are listed in table 3. A total of 406 adverse events were observed. Of these, n = 52 (12.8 %) were considered severe. A total of 238 vs. 168 AEs occurred in the everolimus and tacrolimus group, respectively (χ2 (1df) = 12,069; p = 0.001). There was no difference in the incidence of SAEs in the everolimus and tacrolimus group: 25 vs. 26 (χ2 (1df) = 0.020; p = 0.89). Peripheral oedema and oral ulcers occurred more frequently among everolimus-treated patients. The incidence of all other AEs was not significantly different between the two groups. A dropout rate of 60% (n = 18) was observed in the everolimus group. Of the patients, 30% were reconverted to tacrolimus because of acute rejection (n = 9). Another 26.7% were reconverted because of toxicity (n = 8). Of these eight patients, three were reconverted to tacrolimus because of severe peripheral oedema, one because of peripheral oedema in combination with leucopenia and exanthema, one because of oral ulcers and peripheral oedema, one because of exanthema, one because of pancytopenia, and in one case due to severe pneumonitis. One other patient was reconverted to tacrolimus because of the recurrence of his primary kidney disease (granulomatosis with polyangiitis).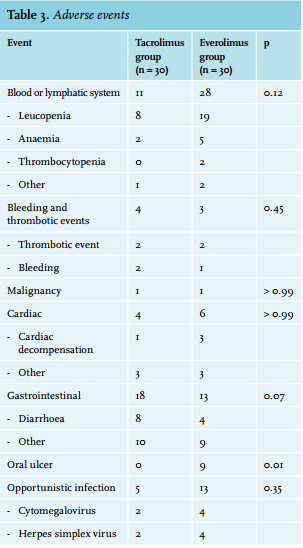
Primary endpoint
At month 12 after transplantation, there was no statistically significant difference with regard to eGFR between the tacrolimus and everolimus group: 53 vs. 56 ml/min per 1.73 m2 , respectively (p = 0.52). The difference at month 12 was 3 ml/min per 1.73 m2 . In the tacrolimus group an average decrease of Δ = -1 ml/min in eGFR occurred over the course of 9 months, whereas in the everolimus group an increase of Δ = 2 ml/min occurred during this same period. Median protein/creatinine ratios were significantly different between the everolimus and tacrolimus group: 18.7 vs. 11.8 mg/mmol, respectively (U = 212.5; p = 0.005; table 4). 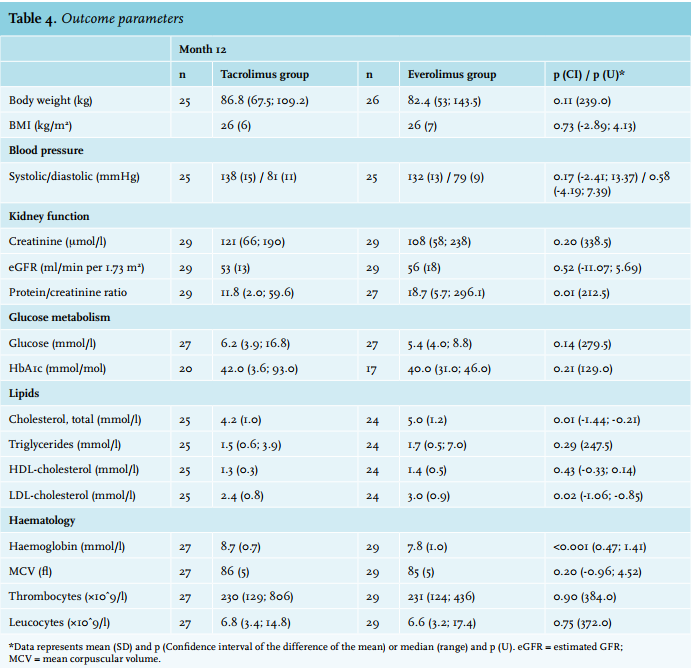
Secondary outcomes
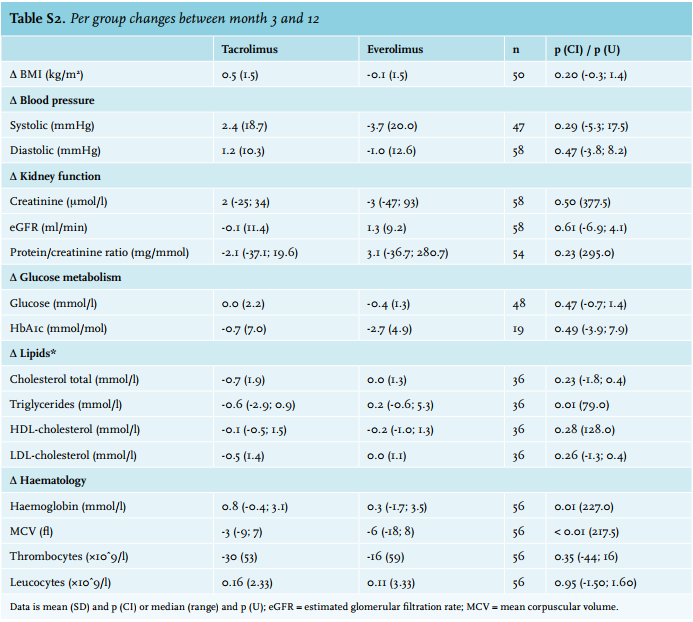
The outcome parameters are listed in table 4. At baseline, no significant differences between efficacy parameters were found. Differences per group between month 3 and month 12 are shown in Supplementary table 2.
At month 12 there was a significant difference in haemoglobin concentration in favour of the tacrolimus group. When adjusted for gender and baseline haemoglobin, the difference between groups remained statistically significantly different (p < 0.001). Total cholesterol level and LDL cholesterol were significantly lower in the tacrolimus group at month 12. All other outcome parameters were not significantly different between the two groups (table 4).
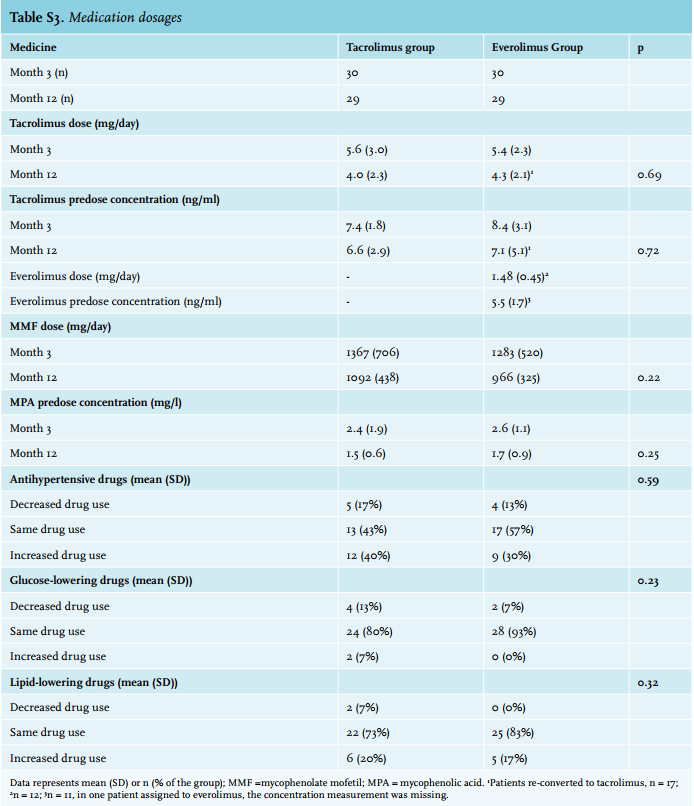
Medication dosages and changes are listed in Supplementary table 3. There were no significant changes in medication usage between groups at month 12. Increased use of antihypertensive and lipid-lowering drugs as compared with baseline was observed in both groups. There were no statistically significant changes in the use of glucose-lowering drugs.
DISCUSSION
Conversion from tacrolimus to everolimus-based immunosuppression three months after transplantation with complete and early withdrawal of glucocorticoids is not safe in living-donor kidney transplant recipients. Conversion results in an unacceptably high risk of acute rejection. Moreover, a considerable number of patients discontinued everolimus because of side effects.
The results of this trial differ from other randomised trials that studied early conversion from a CNI-based to an everolimus-based immunosuppressive regimen.14,15 The investigators of the ZEUS trial concluded that early conversion (month 4.5 after transplantation) from cyclosporine to everolimus resulted in improved kidney function without compromising efficacy and safety.14 In the Dutch multi-centre MECANO trial, renal transplant recipients were converted from cyclosporine, mycophenolate sodium and prednisolone-based immunosuppression to everolimus/prednisolone or cyclosporine/prednisolone combination therapy at month 6 after transplantation. Conversion to everolimus-based immunosuppression resulted in better renal function and better preservation of renal histology compared with patients who were treated with cyclosporine/prednisolonebased therapy.15
In the ELEVATE trial, 715 de novo kidney transplant recipients were randomised at 10-14 weeks to convert to everolimus (n = 359) or remain on standard CNI therapy [n = 356; tacrolimus (n = 231) or cyclosporine (n = 125)] in combination with mycophenolic acid and glucocorticoids.16 In ELEVATE, there was no difference in the primary endpoint, the estimated change in eGFR from randomisation to month 12 post-transplant.16 In line with the observations made in the present trial, in ELEVATE the incidence of BPAR in the everolimus arm (9.7%) was comparable with that of patients who remained on cyclosporine (8.8%) but was significantly higher compared with patients who continued tacrolimusbased immunosuppression (2.6%).16
A major difference between the present study and previous investigations is the complete cessation of glucocorticoids after conversion to everolimus. We chose to eliminate prednisolone in both groups because double immunosuppressive therapy consisting of tacrolimus and mycophenolate mofetil (from month 4 onwards) is standard practice in our centre. In the ZEUS and ELEVATE trials, patients were maintained on ≥ 5 mg of prednisolone (or an equivalent glucocorticoid).14,16 Another difference between the present study and previous investigations was the type of CNI in the control group. In the ELEVATE trial, patients were converted to everolimus from either tacrolimus or cyclosporine, whereas in the ZEUS trial all patients were on cyclosporine at baseline.7,14,16 In the trial reported here, all patients were treated with tacrolimus, which is considered more potent compared with cyclosporine.17
High rates of acute rejection were also observed in other trials in which patients were converted from a CNI to everolimus.7,14,18-20 In the CENTRAL study, 27.5% of the patients randomised from cyclosporine/tacrolimus to everolimus experienced an episode of acute rejection in the first year. CENTRAL had a similar design as the trial reported here, except that the conversion was performed as early as 7 weeks after transplantation.19
Drop out in the everolimus group was high. In 30% of patients, everolimus was stopped because of rejection, whereas in another 26.7% of patients, everolimus was stopped because of toxicity. In most cases, these were typical side effects associated with the use of an mTOR inhibitor, such as oedema and exanthema. Management of side effects was left to the attending physician but an effort was made to keep patients on their assigned treatment. In general, if possible, a dose reduction of everolimus was performed and any concomitant medication (such as co-trimoxazole, valganciclovir, or calcium channel blockers) was first withdrawn or reduced if this was considered the cause of the symptoms. Oral ulcers were often managed with topical steroids. However, several patients requested conversion to tacrolimus and refused further treatment with everolimus when these troublesome side effects occurred.
The high dropout rate because of everolimus-related side effects is consistent with results of other switch studies. However, there is a big difference in discontinuation of everolimus because of toxicity in the first year between studies (12.5%-32.6%).5,6,14-16,19,20 The CENTRAL study investigators reported a 43.1% dropout in the everolimus group in the first year. Of the 43.1% dropout, toxicity accounted for 25.5%, rejection for 13.7%, and other reasons for 3.9%.19 In the ZEUS study, dropout because of toxicity and rejection was 6.5% and 3.9%, respectively, after 1 year in the everolimus group.7 However, 10% of the patients assigned to everolimus experienced an episode of BPAR, indicating that physicians were less inclined to switch back to a CNI, even when treatment with everolimus was failing.7 The willingness of treating physicians to switch back to a CNI after a period of rejection or adverse events may contribute greatly to the differences in dropout observed between studies.
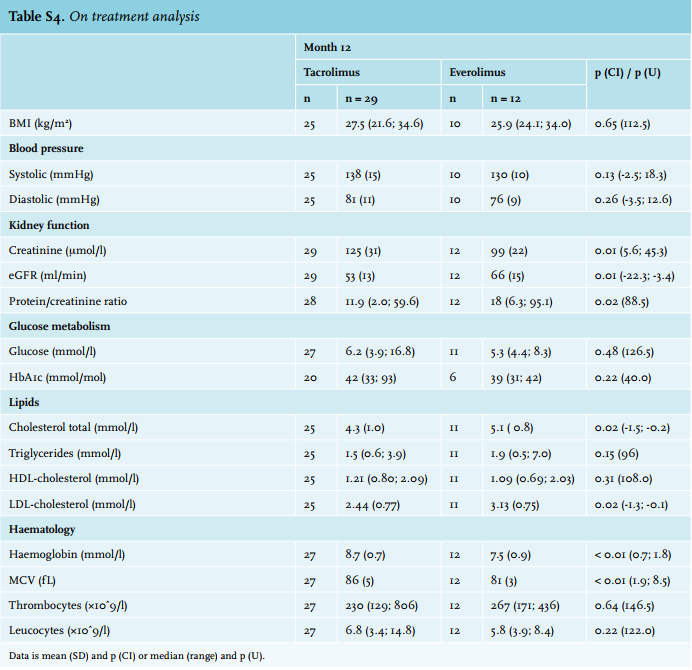
No difference in the primary endpoint was observed between the two groups. At month 12, renal function was comparable between the two groups. Obviously, the present study – which only included a limited number of patients – was not powered to detect such a difference. However, we think it unlikely that any such difference would have been detected if the planned 194 patients had been included. Any benefit of everolimus in terms of improved renal function, as has been reported previously,7-9,15 would likely have been offset by the higher rejection risk. Results from the on-treatment analysis (n = 12; Supplementary table 4) showed a significantly improved kidney function in the group which was switched and continued everolimus vs. those who remained on tacrolimus (eGFR of 66 vs. 53 ml/min, p = 0.01). Thus, a proportion of patients seem to benefit from conversion to everolimus in terms of improved renal function. However, whether this benefit persists over time remains to be determined. Of note, in a long-term follow-up study, patients randomised to everolimus were found to more often develop de novo donor-specific anti-HLA antibodies which is considered a risk factor for chronic rejection.21 Furthermore, there are at present no reliable biomarkers that can assist clinicians in identifying patients who will do well after conversion from a CNI to everolimus.2,22
Our study has several limitations; the trial was ended prematurely which resulted in a small number of patients available for the analysis. The study therefore lacks the power to detect significant differences in the primary endpoint and the small number of patients increases the probability of detecting a difference by chance (type 1 statistical error). However, the significant difference in the incidence of BPAR is in line with other studies and we feel that these results are not random. Second, our population primarily consisted of Caucasian males, making the results not generalisable to all patient populations. Still, as African American transplant recipients are considered high-immunological risk patients, also for this patient group conversion to everolimus may not be advisable.23 Finally, as mentioned above, complete and early cessation of glucocorticoids after month 3 is not standard practice in many transplant centres.
The publication of results of studies that are stopped prematurely is very important.24 These publications provide important information for researchers who are considering to embark on studies with similar goals and study designs. Furthermore, for systematic reviews with meta-analysis, a balanced representation in the literature of studies with positive and negative outcome results is crucial.25
In summary, conversion from tacrolimus to everolimusbased immunosuppression with compete withdrawal of prednisolone three months after living-donor kidney transplantation results in an unacceptably high risk of acute rejection in addition to causing considerable toxicity. Based on the present findings, such a switch strategy should not be considered safe and cannot be recommended.
ACKNOWLEDGEMENTS
The authors are grateful to the research nurses Mrs. M.J. Boer -Verschragen, Ms. M. Cadogan, and Mrs. N.J. de Leeuw -van Weenen for their valuable contribution to this clinical study. The authors would like to thank Mrs. I. Buijt for her help with the statistical analyses. The authors are grateful to Professor H. Boersma, Dr. R.J. Hené, and Professor L.B. Hilbrands for serving in the data safety monitoring board. Dr. Bouamar received a grant (grant number 017006041) from the Netherlands Organization for Scientific Research (NWO; grant number 017006041). Dr. Shuker received a grant (grant number IP11.44) from the Dutch Kidney Foundation (Nierstichting Nederland). All authors had full access to all the data and have full responsibility for the contents of this publication and the decision to submit for publication.
DISCLOSURES
Prof. dr. T. van Gelder has received lecture fees from Chiesi Pharmaceuticals and Astellas Pharma B.V., and consulting fees from Astellas Pharma B.V., Novartis Pharma B.V., Roche Pharma, Teva Pharma and Sandoz Pharma. Dr. D.A. Hesselink has received lecture and consulting fees, as well as grant support from Astellas Pharma B.V., Bristol-Myers Squibb, Chiesi Pharmaceuticals, MSD Pharmaceuticals, Novartis Pharma B.V., and Roche Pharma. The other authors have no conflicts of interest to disclose.
FUNDING
This was an investigator-initiated study. The trial was financially supported by Novartis Pharma B.V., Arnhem, the Netherlands, the producer of everolimus (Certican®). Novartis Pharma B.V. had no role in the study design, data collection, data analyses, data interpretation, or writing of the report. All authors had full access to all the data, had final responsibility for the contents of this publication and the decision to submit for publication.
REFERENCES