KEYWORDS
Kikuchi disease, haemophagocytic syndrome, nephrotic syndrome, liver cell necrosis
INTRODUCTION
Kikuchi disease, also known as Kikuchi-Fujimoto disease or histiocytic necrotising lymphadenitis, was first described in Japan in 1972. The pathogenesis of the disease is unknown, but an immune-mediated response of T-lymphocytes and histiocytes to an as yet unspecified infectious agent is suggested. Kikuchi disease is a rare, benign, self-limiting condition characterised by fever and especially cervical lymphadenopathy. Additionally there is a wide variety of other symptoms and manifestations of the disease that usually recover within one to four months without treatment.1-8
We present a well-documented unique case of a patient suffering from Kikuchi disease with serious multi-organ failure, including nephrotic syndrome, liver cell necrosis and haemophagocytic syndrome. With this case report we aim to enlarge the clinical picture of this rare disease, since nephrotic syndrome in Kikuchi disease has never been reported. Despite the disseminated presentation of the disease, this patient recovered fully without any corticosteroid, immunoglobulin or etoposide treatment.
CASE REPORT
A 54-year-old Caribbean woman, with an unremarkable medical history, was admitted to our hospital because of a six-day history of fever (up to 40°C), cold chills and night sweats. She also complained of generalised joint stiffness, without any signs of arthritis. She received antibiotic treatment (amoxicillin and clavulanic acid), which was not effective. She reported diarrhoea, without any blood or mucus or abdominal pain. She did not report any unprotected sexual contacts or contact with domestic animals.
On admission, physical examination was unremarkable, except for a temperature of 39.7°C and a tachycardia of 116 beats/min. There was no palpable lymphadenopathy and there were no clues for an infectious focus. Laboratory results on admission are shown in figure 1. Chest radiography and abdominal ultrasound were normal except for hepatic steatosis.
During admission all urine cultures, repeated blood cultures and faecal analysis for Salmonella, Shigella, Campylobacter, Yersinia, Shiga toxin-producing Escherichia coli and Clostridium difficile toxin were negative. Serology for syphilis, hepatitis B, C and E, human immunodeficiency virus, Parvovirus B19, toxoplasma, Brucella and Leishmania was negative and showed primary infections with Cytomegalovirus, Epstein-Barr virus and hepatitis A in the past. Testing for systemic autoimmune disease, including antinuclear antibody (weakly positive, with low anti-extractable nuclear antigens), rheumatoid factor 11 kU/l (normal < 20 kU/l) and anti-CCP < 7U/ml (normal < 10 U/ml), was unremarkable.
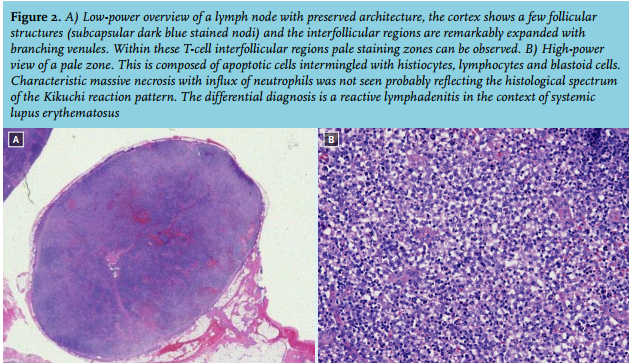
A PET-CT scan, performed eight days after admission because of persistent fever, showed pathological cervical, supraclavicular, axillar, mediastinal and inguinal lymph nodes with sizes that varied from 7 x 15 mm to 7 x 22 mm and a maximum standardised uptake value of 8.9. Histological examination of a cervical lymph node showed reactive lymphadenitis most probably of viral origin without signs of malignant lymphoma (figure 2).
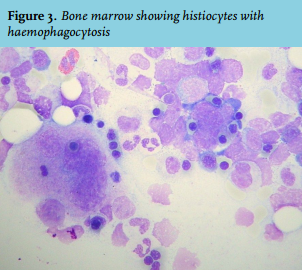
On day 18 after admission, the patient developed nephrotic syndrome with peripheral oedema and rapidly rising liver enzymes (figure 1). There was no hypotension or oliguria and diuresis at that time varied from 1650 ml to 2700 ml per day. Minimal change disease could not be confirmed because electron microscopy was not performed. Renal biopsy showed acute tubular necrosis and podocyte damage. Liver biopsy showed cellular necrosis. Additionally, haemophagocytic syndrome with hyperferritinaemia, hypertriglyceridaemia and extensive haemophagocytosis in the bone marrow was diagnosed (figure 3). At this point, treatment with steroids was postponed because of spontaneous improvement of the clinical and laboratory parameters.
Expert haematopathology panel consultation with additional staining of a cervical lymph node resulted in the diagnosis of Kikuchi disease four weeks after admission. Before immunosuppressive therapy such as corticosteroids, immunoglobulins or etoposide was initiated, the patient improved and recovered fully. During follow-up no residual organ damage was observed.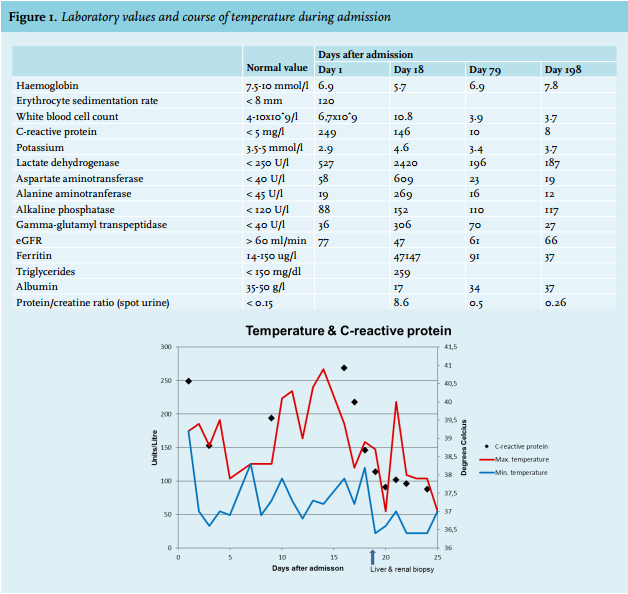
DISCUSSION
In this case report we present a woman with an impressive presentation of Kikuchi disease with multi-organ failure, including renal insufficiency and nephrotic syndrome due to podocyte damage, liver cell necrosis and haemophagocytic syndrome. She recovered completely without any treatment. So far, the combination of renal failure with nephrotic syndrome in the context of Kikuchi disease and the combination of three serious organ manifestations in one patient has not been reported in the literature.
Kikuchi disease is predominantly observed in young females and is primarily characterised by fever and cervical lymphadenopathy. Other frequently observed clinical features are erythematous rashes, arthritis, fatigue, night sweats, diarrhoea, weight loss, anaemia, leukopenia, hepatosplenomegaly and a high erythrocyte sedimentation. Liver failure has been reported in the literature. In accordance with our case, liver biopsies in cases of Kikuchi disease associated with liver failure showed liver cell necrosis.1,3,6,9
As illustrated in our case, enhanced activation of histiocytes can even result in acute renal failure caused by acute tubular necrosis with nephrotic syndrome due to podocyte damage. This renal complication in the course of Kikuchi disease, maybe due to the concomitant haemophagocytic syndrome, is extremely rare. Indeed the combination of nephrotic syndrome in the context of haemophagocytic syndrome has been reported in the past.10 We only found one case of Kikuchi disease with renal failure, but in this case no classifying renal diagnosis was reported.11 Several other rare manifestations of Kikuchi disease have been described, such as aseptic meningitis, cerebellar symptoms and haemophagocytic syndrome. Haemophagocytic syndrome in the context of Kikuchi disease has been documented in 20 case reports until now.12-31
Haemophagocytic syndrome is a rare syndrome of pathological immune activation characterised by clinical signs and symptoms of extreme inflammation. Proliferation of the haemophagocytic cells of the monocytemacrophage-histiocyte lineage results in uncontrolled phagocytosis of normal haematopoetic cells. It is proposed that Kikuchi disease and haemophagocytic syndrome are part of a disease continuum rather than representing separate entities.16,22 This hypothesis originates from the fact that both diseases are associated with marked activation of lymphocytes and histiocytes.28 Most of the patients described with the combination of Kikuchi disease and haemophagocytic syndrome are diagnosed in Asian countries and were children or young adults, with a mean age of 16 years.12,15-23,25,26,29-31
The pathogenesis of Kikuchi disease is still unknown. An immune-mediated response of T-lymphocytes and histiocytes to an as yet unspecified infectious agent is suggested. Examples include Epstein-Barr virus, HIV, parvovirus B19 and human herpes virus 6 and 8.1,3,4,6 There is also an association with systemic lupus erythematosus, since both diseases share histological features. In our case, no underlying provoking infectious agent or disease could be detected and during one year of follow-up, no systemic or rheumatic disease was observed. We can only speculate why our patient developed this serious multi-organ presentation of Kikuchi disease. Compared with other Kikuchi patients with haemophagocytic syndrome, our patient was relatively old. One might hypothesise that in our patient a more profound pro-inflammatory response occurred, mediated by cytotoxic T-lymphocytes and histiocytes resulting in apoptotic cell death in several organ systems due to decreased numbers of regulatory T-lymphocytes, which are essential for immune homeostasis.
The treatment of Kikuchi disease with a variety of clinical characteristics is challenging, especially if complicated by serious multi-organ problems as described in this case report. Because Kikuchi disease is self-limiting in the majority of patients, only supportive care seems to be required. Patients with severe or persisting symptoms can be treated with glucocorticoids or immunoglobulins.32,33
In patients with haemophagocytic syndrome, etoposidecontaining regimens can be considered.34 In our case, we have elegantly shown that an accurate diagnostic approach, including collecting biopsies from the organs involved, can allow potentially toxic therapy to be withheld from the patient. In the literature only a few patients with haemophagocytic syndrome were not treated with corticosteroids, immunoglobulins or etoposide.17,18,21,28
In summary, we present a 54-year-old woman with a disseminated presentation of Kikuchi disease, complicated by nephrotic syndrome, liver cell necrosis and haemophagocytic syndrome, who fully recovered without any immunosuppressive treatment. Although this combination is extremely rare, awareness for early recognition is mandatory and with this case report we enlarge the clinical picture of Kikuchi disease. A thoroughly diagnostic approach can lead to the diagnosis of Kikuchi disease and thus may prevent longstanding immunosuppressive therapy or unnecessary aggressive therapy with cytotoxic agents.
DISCLOSURES
The authors declare no conflict of interest. No funding or financial support was received.
REFERENCES