KEYWORDS
Ferric carboxymaltose, fibroblast growth factor (FGF)-23, hypophosphataemia, iron, kidney transplantation, phosphate.
INTRODUCTION
Ferric carboxymaltose (FCM) is a non-dextran iron complex that is composed of a ferric hydroxide core stabilised by a carbohydrate shell, carboxymaltose, allowing controlled delivery of iron to the target tissues.1 FCM is indicated for the treatment of iron-deficiency anaemia when oral iron preparations are either ineffective or not tolerated. The drug allows rapid and convenient repletion of iron stores and is generally well tolerated. Apart from rare anaphylactic reactions, adverse drug reactions are mild and include nausea (reported in approximately 7% of patients), hypertension (4%), flushing (3.5%), dizziness (2%), and vomiting (1.5%).1-3
Hypophosphataemia is another side effect of FCM.1 Transient decreases in serum phosphate occur in 3.7-58.8% of the general population and are mostly asymptomatic.1,4,7-10 Hypophosphataemia also occurs following intravenous administration of saccharated ferric oxide and iron polymaltose.4-6,8 The risk of developing hypophosphataemia appears to be greater with FCM as compared with other intravenous iron preparations.4,7-9 A decrease in phosphate concentration may be a reflection of cellular uptake of extracellular phosphate associated with a rapid expansion of erythropoiesis.11 However, fibroblast growth factor (FGF)-23 seems to be a more important factor in the hypophosphataemia induced by intravenous iron.9,10 Studies in iron-deficient women have demonstrated that parenteral iron reduces kidney phosphate reabsorption, promotes phosphaturia, decreases calcitriol concentration and that this is mediated by an increased concentration of FGF-23. FCM may reduce the cleavage of intact and biologically active FGF-23 (iFGF-23) after it is secreted by osteocytes.9,10
In contrast to patients receiving intravenous iron to correct iron deficiency caused by menstrual blood loss, inflammatory bowel disease, or bariatric surgery, chronic kidney disease (CKD) is characterised by unique, chronic, and profound alterations in phosphate and vitamin D metabolism. FGF-23 plays an important role in phosphate homeostasis and vitamin D metabolism. It induces phosphaturia and suppresses calcitriol synthesis.12-15 In CKD and haemodialysis patients, hyperphosphataemia rather than hypophosphataemia is the rule and relates to a reduced renal capacity to excrete dietary phosphate loads leading to increased concentrations of FGF-23. Blood concentrations of FGF-23 increase as kidney function declines, with haemodialysis patients having the highest concentrations.12,13,16 In CKD patients (both non-dialysis dependent and haemodialysis-dependent) the reported incidence of FCM-induced hypophosphataemia varies between 3.8% and 75%.17-19
Following successful kidney transplantation, some of the abnormalities in phosphate and vitamin D metabolism seen in CKD persist despite normalisation of kidney function. In many kidney transplant recipients, calcitriol concentrations remain low despite good graft function and hypophosphataemia occurs frequently. Several studies have suggested that persistent increases in plasma FGF-23 concentrations rather than tertiary hyperparathyroidism cause post-transplant hypophosphataemia.15,20
Only limited information on the safety of FCM in the kidney transplant population is available. At present, two cases of severe FCM-induced hypophosphataemia (necessitating hospital admission) after kidney transplantation have been reported. Only a single FGF-23 concentration was measured in one of these patients and found to be elevated.21,22 In the current study, we describe a series of kidney transplant recipients developing hypophosphataemia after intravenous FCM administration. The aim of this study was to investigate the incidence and severity of hypophosphataemia following a single dose of intravenously administered FCM after kidney transplantation. Our observations suggest that FGF-23-mediated kidney phosphate wasting is the most likely cause of this phenomenon.
METHODS
Patients
In the Erasmus MC, University Medical Center Rotterdam, the Netherlands, approximately 200 kidney transplantations are performed each year. The number of kidney transplant recipients followed at our outpatient clinic is approximately 1900.
FCM (Ferinject®, Vifor Pharma Ltd., Switzerland) was approved to treat iron-deficiency anaemia in the Netherlands on 6 July 2007 and has been the iron preparation of choice in our unit since the spring of 2011. Two kidney transplant patients received FCM in 2013 and subsequently developed hypophosphataemia. In these index patients more extensive measurements related to phosphate homeostasis (including FGF-23) were performed and these two patients are described first. These observations led us to further analyse the relationship between the administration of a single dose of FCM and changes in the serum phosphate concentration. We retrospectively collected data of all kidney transplant recipients who received FCM intravenously between 1 January 2014 and 1 July 2015. Patients who were treated with FCM were identified by means of the electronic prescription and medication distribution system of the Erasmus MC through the department of hospital pharmacy. The two index patients were not included in this analysis.
In order to be included in this case series patients had to have 1) a functioning kidney transplant and 2) at least one serum phosphate concentration measured within three months after FCM administration. Kidney transplant recipients suffering from delayed graft function or a failed transplant and receiving dialysis treatment were not considered.
For all patients the following data were collected, if available: age, gender, primary kidney disease, date of transplantation, time after transplantation, kidney function and serum phosphate, calcium, calcidiol, calcitriol and parathyroid hormone (PTH) concentration.
Chemical analysis
Plasma creatinine and phosphate concentrations were measured as part of routine clinical care at the department of clinical chemistry of our hospital by the enzymatic creatinine23 and Molybdenum blue assays,24 respectively. Estimated GFR (eGFR) was determined by means of the CKD Epidemiology Collaboration (CKD-EPI) study equation.25 The fractional excretion of phosphate was calculated by multiplying phosphate in urine with creatinine in serum, divided by the multiplication of phosphate in serum with creatinine in urine, which was then multiplied by 100. PTH was measured by enzyme-linked immunoassay (Vitros ECi).26,27
Calcidiol and calcitriol were measured by means of a radioimmunoassay (DiaSorin and IDS, respectively).28,29 C-terminal FGF-23 (cFGF-23) was determined in EDTA plasma using the cFGF-23 immunoassay (Immunotopics), which measures both intact (and biologically active) FGF-23 (iFGF-23), as well as (inactive) C-terminal fragments of FGF-23.30 FGF-23 was measured in the fasting state. Because no assay to measure FGF-23 was operational in our hospital at the time of writing, it was measured by means of a validated assay in an external laboratory (Department of Clinical Chemistry, VU Medical Center).
Statistical analyses
Data are presented as mean ± standard deviation or median and range, depending on the distribution of the data. For comparison of variables before and after FCM administration, a paired t-test was used. Spearman’s rho was performed for rank correlation. The Mann-Whitney test and Chi-square with Yates’ correction were used to compare groups. A p value < 0.05 was considered statistically significant.
RESULTS
Index patients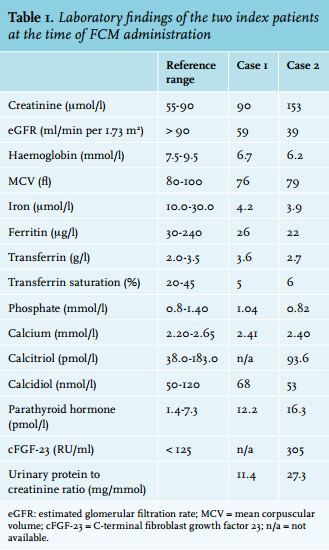
Case 1
A 42-year-old female of Indian descent received a first, pre-emptive, living-related donor, kidney transplant in May 2012 because of end-stage renal disease (ESRD) caused by hypertensive nephropathy. Some seven months after transplantation, iron-deficiency anaemia was diagnosed and considered to be the result of menstrual blood loss in combination with a strict vegetarian diet. She was started on oral iron supplements. Because of persistent iron-deficiency anaemia, she was treated with a single dose of 1000 mg FCM intravenously. At the time of iron infusion, she was treated with tacrolimus and mycophenolate mofetil (MMF). Her other medication consisted of ferrogluconate (695 mg/day), colecalciferol (25,000 IU/month) and amlodipine. Laboratory findings are listed in table 1.
Two weeks after an uneventful administration of FCM, her kidney function remained excellent (eGFR 67 ml/min per 1.73 mm2 ) and iron stores, haemoglobin and MCV had all normalised. However, considerable hypophosphataemia was noted (0.33 mmol/l; figure 1A). This was considered to be the result of renal phosphate wasting as the fractional excretion of phosphate was 34.1% (reference range < 5%) with an absolute urinary phosphate loss of ~1.4 g/day. She had no glucosuria, urinary pH was normal at 6, and there was no evidence for a plasma cell dyscrasia, arguing against a generalised tubular defect (aminoaciduria and plasma bicarbonate were not determined). The calcium concentration was 2.30 mmol/l, calcidiol and calcitriol concentrations were within the normal range (66 nmol/l and 100 pmol/l respectively). PTH had increased slightly (14.5 pmol/l). The cFGF-23 concentration was elevated at 160 RU/ml (reference range < 125).
The patient did not receive any additional treatment. Two months after intravenous FCM administration, the serum phosphate had normalised to 0.80 mmol/l. The PTH concentration had returned to baseline (12 pmol/l), calcidiol remained within the reference range (53 nmol/l) and calcitriol concentration had increased, although it remained within the reference range (168 pmol/l). cFGF-23 decreased to the reference range (106 RU/ml).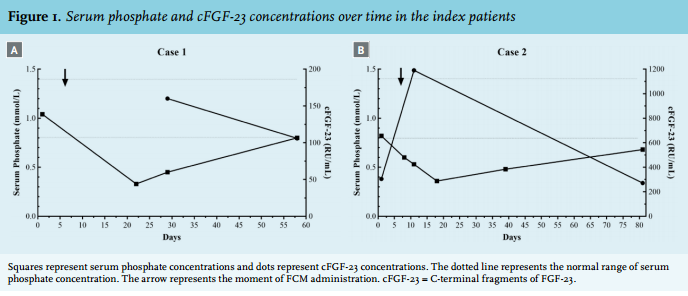
Case 2
A 70-year-old Caucasian male received his first, pre-emptive, living-unrelated donor, kidney transplant in March 2013 because of ESRD caused by hypertensive nephropathy, possible glomerulonephritis in the 1940s, and nephrolithiasis. In addition, he underwent a right-sided hemi-colectomy three years prior to transplantation because of adenocarcinoma of the ascending colon. Some five months after transplantation, iron-deficiency anaemia was diagnosed and considered to be caused by chronic blood loss due to a benign ulceration at the site of the neo-ileo-colonic anastomosis. He received a single dose of 1000 mg FCM intravenously followed by oral iron supplementation for the duration of one month. Some nine months after transplantation and approximately three months after his first FCM administration, recurrent iron-deficiency anaemia was diagnosed. It was decided to treat the patient with a second administration of 1000 mg FCM. At this time, he was treated with tacrolimus, MMF, prednisolone, esomeprazole, trimethoprim/ sulfamethoxazole, amlodipine and acenocoumarol. In addition, he received 0.25 μg of alphacalcidol because of persistent hyperparathyroidism (PTH 16 pmol/l). Laboratory results are listed in table 1.
Three days after an uneventful FCM infusion, the serum phosphate concentration had dropped to 0.53 mmol/l and decreased further to a nadir of 0.36 mmol/l ten days after FCM infusion (figure 1B). There was marked renal phosphate wasting with a fractional excretion of phosphate of 52.7% and 75.1% three and ten days after infusion, respectively. The FGF-23 concentration increased up to 1190 RU/ml three days after the infusion. Kidney function remained stable at 43 ml/min per 1.73 m2. The calcium concentration was 2.32 mmol/l. The calcidiol concentration was within the normal range (57 nmol/l), but the calcitriol concentration declined significantly (57 pmol/l). PTH concentration had increased to 19 pmol/l. There was no glucosuria or proteinuria on urinalysis.
The patient did not receive additional treatment. Two and half months after the second FMC administration, serum phosphate concentration had increased to 0.68 mmol/l. The PTH and calcitriol concentration had returned to baseline (16 pmol/l and 98 pmol/l), while calcidiol remained normal (52 nmol/l). The fractional excretion of phosphate and cFGF-23 concentration decreased to 19.3% and 271 RU/ml, respectively.
Additional kidney transplant recipients
A total of 30 kidney transplant recipients who received FCM intravenously between 1 January 2014 and 1 July 2015 were identified. Two of these patients were excluded due to the absence of phosphate concentration measurements after FCM administration. Four patients were excluded because they had delayed graft function and were treated with haemodialysis when they received FCM. One patient experienced kidney allograft failure and was started on haemodialysis shortly before receiving FCM. Therefore, a total of 23 kidney transplant recipients were included in this case series. The characteristics of these patients are summarised in table 2. The two index patients were not included in this analysis. Only limited laboratory results for PTH, calcitriol and calcidiol were available for these patients.
The median serum creatinine concentration at the time of FCM administration was 200 μmol/l and ranged from 61 to 529 μmol/l with a median eGFR of 42 ml/min/1.73 m2 (range of 10 to 90 ml/min per 1.73 m2 ). One patient was treated with 100 mg FCM intravenously, 3 with 500 mg and 19 with 1000 mg.
As can be seen from table 2, serum phosphate concentrations were within the reference range in 11 (47.8%) patients before FCM administration. In nine patients (39.1%) serum phosphate concentrations were decreased and in three patients (13.0%) elevated at the time of FCM treatment. The median time to the first phosphate measurement after a single dose of FCM was 20 days (range 3 to 53 days).
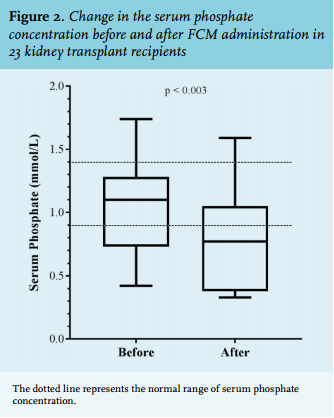
Thirteen patients (56.5%) developed hypophosphataemia after FCM administration. The mean serum phosphate concentration decreased by an average of 0.27 mmol/l from 1.05 ± 0.35 mmol/l (before FCM) to 0.78 ± 0.41 mmol/l (after FCM administration); p = 0.003 (figure 2). In eight (34.8%) patients the hypophosphataemia was severe (defined as < 0.5 mmol/l). The median time to hypophosphataemia was 15 days (range 3 to 24 days). In the three months following the diagnosis of hypophosphataemia, serum phosphate concentrations normalised in eight patients (61.5%), whereas hypophosphataemia persisted in five patients (38.5%). The median time to normalisation of serum phosphate concentrations was 41 days (range 2 to 99 days). From the retrospective patient chart review, none of the patients seemed to have experienced additional side effects of FCM. In one case, phosphate supplementation was started. Of the total population, five patients (21.7%) showed a transient reduction in phosphate concentration without the development of hypophosphataemia. Calcium concentrations were measured in 22 patients. The mean serum calcium concentration did not change significantly: 2.32 ± 0.25 mmol/l (before FCM) vs. 2.30 ± 0.16 mmol/l (after FCM); p = 0.56.
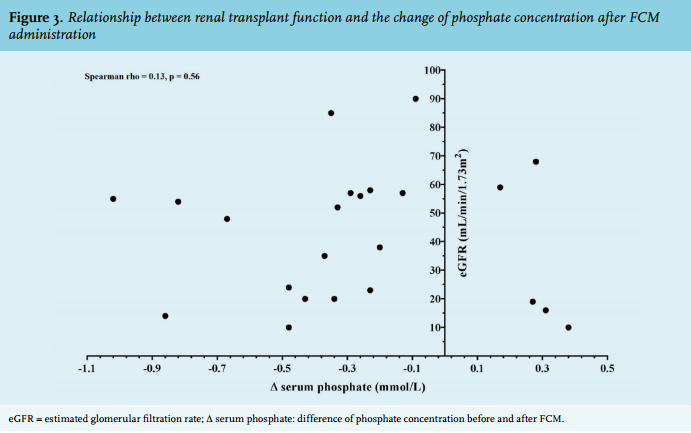
The relationship between renal transplant function and the change of phosphate concentration after FCM administration was analysed (figure 3). The delta phosphate concentration (difference before and after FCM) were not correlated with the renal transplant function (Spearman rho = 0.13, p = 0.56).
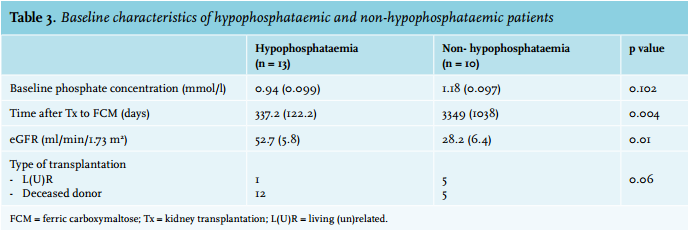
Next, potential risk factors for the development of hypophosphataemia following FCM infusion were investigated. The change in serum phosphate concentrations (i.e. the difference before and after FCM administration) was analysed in relation to the baseline phosphate level (at the time of FCM administration), renal function, time after transplantation, and type of donor (living vs. deceased donor). These results are described in table 3. Taken together, patients who developed hypophosphataemia following FCM treatment had a shorter time after transplantation and had better renal function compared with patients who did not develop hypophosphataemia. Baseline serum phosphate concentration and type of donor were not different between patients with and without hypophosphataemia.
DISCUSSION
Kidney transplantation often only partially reverses the profound alterations in calcium, phosphate, and vitamin D metabolism that characterise CKD. Many kidney transplant recipients will develop a ‘tertiary hyperphosphatonism’ with persistently elevated FGF-23 concentrations, decreased calcitriol concentrations, and inappropriate kidney phosphate wasting resulting in hypophosphataemia.15 It is believed that the bones are the source of the excessive FGF-23 production but it is unclear why they continue to release FGF-23 after transplantation.15,31 Although tertiary hyperparathyroidism may contribute, FGF-23 may be the most important determinant of post-transplant hypophosphataemia.15,20,31 In case 2 of our series, FGF-23 concentrations remained elevated ten months after kidney transplantation.
Previous studies reported an increase in FGF-23 concentrations after intravenous iron supplementation in non-CKD patients with iron-deficiency anaemia. This was followed by a transient and asymptomatic reduction in serum phosphate.6,9,11,32 Likewise, in non-dialysis dependent CKD patients and in patients on haemodialysis, a transient and asymptomatic decline in serum phosphate concentration after FCM administration has been reported which may persist for up to three months.17-19,33 In CKD patients, FGF-23 concentrations are often elevated and FCM administration seems to further increase these concentrations. Our observations are in line with these findings and suggest that FCM also induces kidney phosphate wasting with hypophosphataemia after kidney transplantation and that this is likely mediated by FGF-23. In our population, 56.5% of the patients developed hypophosphataemia after a single dose of FCM. In the two patients in whom FGF-23 was measured, increased concentrations of FGF-23 were observed after FCM administration followed by a decline that was mirrored by normalisation of serum phosphate concentrations. The increase in PTH and decrease of calcitriol concentrations can also be explained by increased FGF-23 activity.
Only few data exist in the literature regarding the risk of kidney transplant recipients to develop hypophosphataemia after FCM therapy. Mani et al.21 and Blazevic et al.22 each reported a case of FCM-induced hypophosphataemia after kidney transplantation. The series reported here describes the largest cohort of kidney transplant recipients developing a reduction of serum phosphate concentration after FCM administration and the index cases point towards an important role of FGF-23 in the pathophysiology. Our observation that patients who developed hypophosphataemia after FCM treatment were more recently transplanted and had a better renal function is in line with this hypothesis. Following a successful kidney transplantation, the inability to induce phosphaturia may be reduced and the nephrons may be more sensitive to FGF-23. In a good transplant function it may lead to hypophosphataemia when FGF-23 concentrations are high.
Both iron infusion and iron deficiency appear to stimulate FGF-23 production by osteocytes. Recent studies suggest that iron-deficiency anaemia is associated with normal concentrations of hormonally active, intact FGF-23 (iFGF-23) but markedly elevated inactive cFGF-23 concentrations without influence on the phosphate metabolism.9,34,35 FCM may increase circulating concentrations of iFGF-23 by inhibiting its cleavage within osteocytes into cFGF-23. Although the mechanism remains unclear, it seems that FCM disrupts the balance between FGF-23 production and cleavage within osteocytes.9,11,34,35
The patients described in the current study received a variety of single doses of FCM. Of note, also the one patient who received a single 100 mg FCM dose developed hypophosphataemia. In one of the three patients receiving 500 mg FCM, the phosphate concentration decreased below the reference value. Observations made in the FIND-CKD study36 suggest that the risk of reduction in phosphate concentration after FCM is dose-dependent. Apart from being dose-dependent, the risk of developing hypophosphataemia appears to be higher when FCM is prescribed as compared with other intravenous iron preparations.17-19,33
It is unknown whether persistent hypophosphataemia after kidney transplantation is merely a laboratory peculiarity or whether it has clinical implications. Hypophosphataemia after kidney transplantation is usually asymptomatic and frequently resolves with time although it may persist for more than a year after transplantation.20,37
It may be that chronic kidney phosphate wasting, in addition to traditional risk factors such as the use of glucocorticoids, contributes to the increased fracture risk of kidney transplant recipients.38-40 In our patients, FCM-induced hypophosphataemia did not cause clinical symptoms and resolved spontaneously. However, this was a retrospective analysis and not a prospective study. Therefore mild symptoms and complications following FCM administration may not have been recorded in the patient files. Reports from the literature have indeed suggested that FCM-induced hypophosphataemia may not always be harmless and may necessitate hospital admission and intravenous phosphate supplementation.21,22,41 In addition, repeated administration of FCM, leading to chronic phosphate wasting, might cause osteomalacia.
It is not clear how to manage post-transplant hypophosphataemia. In analogy to CKD, vitamin D is often prescribed, especially when PTH concentrations are elevated. However, calcitriol stimulates FGF-23 secretion and may thus maintain renal phosphate wasting. Likewise, phosphate supplements may induce FGF-23 and could also increase the risk of kidney calcium-phosphate depositions and kidney stones. Frequent administration of FCM in kidney transplant recipients might further increase this risk. In such cases, close monitoring of phosphate concentrations and prudent use of FCM seems warranted. Our study has several limitations. First of all, this was a retrospective analysis and the number of cases is relatively small; FGF-23 concentrations were only measured in two index patients. Second, blood sampling following FCM administration was not standardised. As a result, hypophosphataemia (or its nadir) may have been missed in some patients. Finally, both active iFGF-23 and cFGF-23 were measured by the C-terminal FGF-23 assay we used. This precludes a deeper understanding of the pathophysiology of FCM-induced hypophosphataemia and the role of intact FGF-23 and its fragments therein.
CONCLUSION
Correction of iron-deficiency anaemia after kidney transplantation with FCM may induce renal phosphate wasting and hypophosphataemia in as many as 56.5% of patients. This phenomenon may be explained by increased concentrations of FGF-23. The hypophosphataemia following a single dose of FCM administration was transient. Nonetheless, prudent use of FCM seems warranted and close monitoring of such patients seems advisable, especially when chronic therapy with FCM is indicated.
DISCLOSURES
The authors declare that they have no conflict of interest and no relevant financial interests.
REFERENCES