KEYWORDS
Calcium channel antagonist, calcium channel blocker, hyperinsulinaemia/euglycaemia therapy, intoxication, overdose, poisoning
INTRODUCTION
Calcium channel antagonists (CCAs) are widely prescribed for a variety of indications, e.g. hypertension, coronary artery disease and specific cardiac arrhythmias.1,2 In general, CCAs have a favourable adverse effect profile. However, CCA overdose is common and associated with a relatively high degree of morbidity and mortality.1,3 In the Netherlands, 158 CCA exposures (∼50% intentional) were reported to the Dutch Poisons Information Centre in 2014. Adults (18-65 years) were mostly involved (48%), followed by the elderly (> 65 years, 27%) and young children (0-4 years, 20%). Within the group of cardiovascular drugs, CCAs are the leading class of drugs associated with poisoning fatality.3 In this review, we will discuss the most relevant treatment options for CCA-overdosed patients.
MECHANISM OF ACTION
Three main classes of CCAs are currently on the market: dihydropyridines (e.g. nifedipine, amlodipine), phenylalkylamines (verapamil) and benzothiazepines (diltiazem).1,2,4,5 At therapeutic doses, the dihydropyridines predominantly affect vascular smooth muscle cells and show little effect on the myocardium, making this class of CCAs particularly attractive in treating hypertension. In contrast, the non-dihydropyridine CCAs (verapamil, diltiazem) have the strongest affinity to the myocardium2,4,6 and are used to treat angina or cardiac arrhythmias.
The primary mechanism of action of CCAs is inhibiting calcium influx by antagonism of the L-type voltage-gated calcium channels in vascular smooth muscle cells and cardiomyocytes of the working myocardium as well as the conduction system.7 Blockage of L-type calcium channels in the myocardium results in decreased myocardial contractility, decreased heart rate, decreased excitability and decreased conductivity within the heart. Antagonism of vascular L-type calcium channels results in relaxation of vascular smooth muscle cells, increasing coronary vascular dilatation and decreasing systemic blood pressure.2,6,7
PHARMACO KINETICS
Most CCAs are rapidly absorbed from the gastrointestinal tract (peak concentration ∼1-2 h). Some CCAs (e.g. amlodipine) and also sustained-release preparations show a much slower absorption (peak concentration within 6-12 h). Many CCAs have a low bioavailability due to extensive hepatic first-pass metabolism. In general, CCAs are highly protein bound and have a large volume of distribution.2,5,8,9 In overdose, peak concentrations can be delayed up to ∼24 hours for sustained-release preparations,8 delaying the onset of intoxication symptoms. Furthermore, the apparent half-life of many CCAs can be increased after overdose, because of saturation of metabolism causing prolonged toxicity.2,5,8
TOXICITY RESULTING FROM CCA OVERDOSE
Cardiovascular toxicity
The observed cardiovascular toxicity after CCA overdose is largely an extension of the therapeutic effects. In general, overdose of diltiazem (benzothiazepine) and verapamil (phenylalkylamine) can cause serious hypotension and bradycardia, which can be aggravated by a diversity of sinoatrial, atrioventricular (AV) and bundle branch conduction disturbances, including complete AV-blockage.6,10,11 In contrast, dihydropyridine CCAs tend to create more prominent hypotension accompanied by reflex tachycardia after overdose.6,10,11 However, in severe poisoning, the selectivity for cardiac versus peripheral vascular effects can be profoundly decreased, making the cardiovascular effects less predictable. Initially, patients may be asymptomatic early after ingestion, but can subsequently deteriorate rapidly towards severe and refractory cardiogenic shock.5
Non-cardiovascular toxicity
Interestingly, various non-cardiovascular effects have been described following CCA overdose, such as confusion, agitation, impaired consciousness and seizures,5,8,11-13 which might be caused by hypoperfusion of the central nervous system. Moreover, in overdose, CCAs may inhibit calcium channels outside the cardiovascular system. Non-selective blockage of L-type channels of pancreatic islet cells decreases insulin release resulting in hyperglycaemia.11,13-17 A metabolic state similar to diabetic ketoacidosis may develop, in addition to lactic acidosis.2,15,18 Lactic acidosis is a manifestation of poor tissue perfusion and can be aggravated by inhibition of mitochondrial calcium entry leading to decreased pyruvate dehydrogenase activity.18,19 Other effects are bowel infarction and ileus.2,20-22 Pulmonary oedema23-26 (both cardiogenic and non-cardiogenic) might be caused by excessive fluid resuscitation and precapillary vasodilatation, resulting in increased transcapillary pressure.26,27 Refractory shock and cardiac arrest may ultimately result in death.
Beta-blocker overdose usually causes similar symptoms, although subtle differences may be present. Dihydropyridine CCA overdose generally causes profound hypotension and reflex tachycardia, rather than bradycardia. Nevertheless, distinguishing dihydropyridine overdose from beta-blocker overdose remains difficult. Glucose concentration is not useful for discriminating between CCA and beta-blocker overdose, as both hypoglycaemia and hyperglycaemia are occasionally reported in beta-blocker overdose.15
TREATMENT OF CCA OVERDOSE
As treatment of CCA overdose can be complicated, consultation with a clinical toxicologist, hospital pharmacist and a Poisons Information Centre is strongly recommended. Several factors influence the response to CCA poisoning: drug class, dose, formulation, time of ingestion, co-exposures (e.g. beta-blockers) and pre-existing diseases.8 Elderly patients and patients with underlying cardiovascular disease are expected to be more vulnerable to the toxic effects of CCAs.6
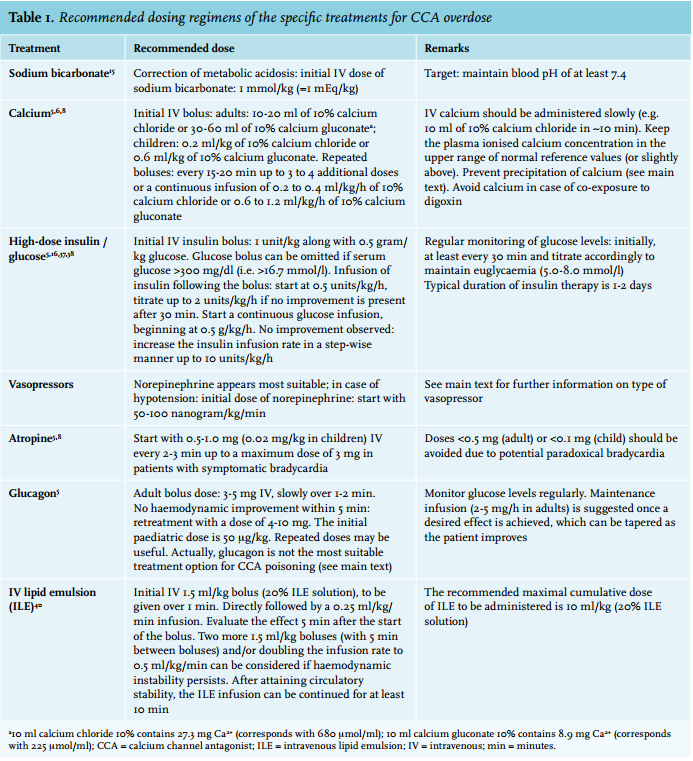
A rational approach to the treatment of CCA overdose is presented in figure 1. Severe cases may require several simultaneous interventions. Recommended doses and possible adverse effects are presented in table 1 and 2, respectively.
In this review, we will provide diagnostic and treatment recommendations for CCA overdose, which are based upon ‘low quality of evidence’. Unfortunately, the current clinical evidence is limited, because it is generally based upon animal studies, case reports and low-quality observational data, which are prone to confounding and biases (see also the review by St-Onge et al.28). Often multiple treatments are simultaneously applied in one patient, making it hard to identify the relative advantages of one intervention over another. 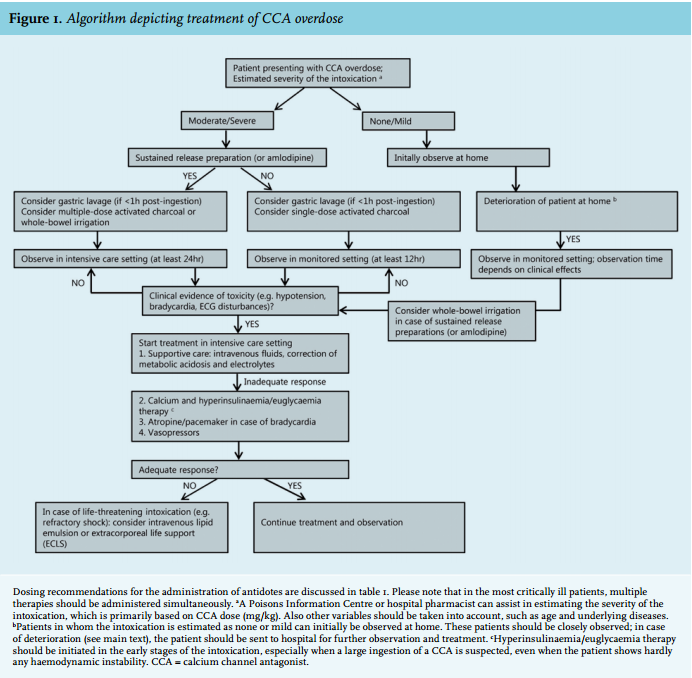

Laboratory testing
Besides routine laboratory testing, an emphasis should be put on blood gas analyses and assessment of lactate, electrolytes, blood glucose and renal function.5,8 CCA serum concentration analyses are not routinely performed and are not usually available on time to guide therapy. In addition, CCA concentrations do not correlate with clinical presentation,5,8,29 although CCA detection above the therapeutic level can indicate an overdose. A general toxicological screening in blood or urine provides information on relevant co-exposures.
Monitoring and supportive care
Close monitoring of vital signs (including electrocardiography (ECG)) in an intensive care setting is fundamental for patients in whom a moderate to severe intoxication is anticipated (based on CCA dose and underlying comorbidities), as rapid changes in clinical condition may occur.1,8,29 Patients should be observed for at least 12 hours if an immediate-release formulation is ingested and at least 24 hours when a sustainedrelease formulation (or amlodipine) is involved, even if asymptomatic.5
Treatment should first be focused on gastrointestinal decontamination and supportive care, e.g. correction of metabolic disturbances and electrolytes. Furthermore, assuring haemodynamic stability and adequate respiration is essential. Acidosis can worsen myocardial dysfunction and the responsiveness to catecholamines,30 which might be due to increased drug binding at the calcium channel, and should be treated by administrating sodium bicarbonate (target of maintaining blood pH of at least 7.4).15 When extensive resuscitation of fluids is needed, norepinephrine should be added. Prevent fluid overload in order to avoid pulmonary oedema.
Gastrointestinal decontamination
Early and aggressive gastrointestinal decontamination is of utmost importance in patients with a potentially toxic ingestion of a CCA. However, gastric lavage should only be considered in patients who present within one hour after ingestion of a large dose. Induced emesis is not recommended as patients could rapidly deteriorate, increasing the risk of aspiration.1,2,5 Activated charcoal can be considered in patients who have ingested a potentially toxic dose,1,2,5 preferably within one to two hours after ingestion (adults: 50 g; children: 1 g/kg, max 50 g). Because of the risk of aspiration, gastric lavage and activated charcoal are contraindicated in patients with unprotected airways, e.g. in patients with depressed consciousness without endotracheal intubation.
Whole-bowel irrigation using polyethylene glycol solution, or multiple doses of activated charcoal should be considered in patients who have ingested a high dose of a sustained-release preparation.1,5,31 Whole-bowel irrigation should be used cautiously in debilitated patients. The potential advantage of reducing CCA absorption should be weighed against the possible disadvantages, e.g. pulmonary aspiration in patients with unprotected airways.
Enhanced elimination
Haemodialysis and haemoperfusion are unlikely to be effective, as CCAs generally have high protein binding and a high volume of distribution.1,2,8,9 Extracorporeal albumin dialysis, using a molecular adsorbent recirculating system that efficiently removes protein-bound toxins from the circulation, may be considered when conventional symptomatic treatment fails.32,33 However, this rescue therapy is not widely available and its efficacy remains controversial.
Calcium
The aim of administrating calcium in CCA overdose is to increase extracellular calcium concentrations, theoretically allowing calcium influx via unblocked L-type calcium channels.5,6 Animal studies generally show positive results, i.e. reduced mortality as well as haemodynamic improvement.28 Case series and case reports in humans are inconsistent,28 probably explained by differences in calcium dose or the severity and the stage of intoxication. Although the positive effects of calcium are often short-lived and more severely intoxicated patients may not improve significantly by administration of calcium alone,5 we recommend the use of calcium as a first-line treatment in CCA overdose, which should be started simultaneously with hyperinsulinaemia/euglycaemia therapy in severe cases.
Calcium should not be co-administered with a sodium bicarbonate infusion because a precipitate is formed.8 Multiple other medications are known to form a precipitate with calcium. ECG monitoring should generally be performed as a safety measure when administrating calcium in order to detect episodes of potentially life-threatening ventricular arrhythmias and conduction abnormalities. When co-intoxication with digoxin is suspected, calcium should be avoided, because this may worsen the digoxin toxicity.5,8
Hyperinsulinaemia/euglycaemia therapy
Hyperinsulinaemia/euglycaemia therapy has become a first-line intervention in CCA-intoxicated patients.1 Failure of this therapy is predominantly reported when it was introduced too late. It, therefore, seems rational to initiate this therapy in the early stages of the intoxication, especially when a large ingestion of a CCA is suspected16,34,35 even when the patient shows hardly any haemodynamic instability. The response to insulin is usually delayed for 15 to 60 minutes. This is an additional argument to start hyperinsulinaemia/euglycaemia therapy early, but alongside alternative treatments.
It is thought that high-dose insulin supports cardiac metabolism during shock states. When cardiomyocytes are ‘stressed’, their metabolism switches from free fatty acids to glucose. In this context, insulin promotes carbohydrate metabolism by increasing myocardial glucose uptake, enhancing inotropic function.15,34,36 High-dose insulin also affects intracellular calcium handling, which contributes to the inotropic effects.34 A large variation in applied insulin dosages has been reported in CCA-overdosed patients. High-dose insulin (IV bolus of 1 unit/kg followed by an infusion of 0.5-2.0 units/kg/h) generally demonstrated positive effects on haemodynamic parameters in humans and animals.28 In the past, lower dosages of insulin were generally used due to concerns about provoking hypoglycaemia. Increased clinical experience and successful treatment of patients using relatively high quantities of insulin resulted in the current dosing strategy typically suggested in the medical literature (table 1). Yet, even more aggressive insulin protocols have been described, suggesting to increase the insulin infusion rate in a step-wise manner up to 10 units/kg/h if no clinical improvement is observed.37 Obvious side effects of high-dose insulin therapy are hypoglycaemia and hypokalaemia (table 2).1,5,16,28,34
Vasopressors
Large doses, as well as the use of multiple vasopressor and inotropic agents, may be required in treating hypotension in severe CCA overdose.1,10,15 No single adrenergic agent has been shown to be superior to another.15 However, norepinephrine appears to be the most suitable initial catecholamine to use in hypotensive CCA-overdosed patients, as it has strong α1-adrenergic activity (to increase peripheral vascular resistance) and moderate β1-adrenergic activity (to enhance myocardial function). α1-Adrenergic agonists (e.g. phenylephrine) seem rational choices if the hypotension is primarily caused by peripheral vasodilation, typically present in patients with dihydropyridine CCA overdose. Theoretically, the use of pure β-adrenergic receptor agonists, such as isoproterenol, and, to a lesser extent, dobutamine, is controversial as this may worsen hypotension mediated by β2-adrenergic receptor agonist-induced peripheral vasodilation.38
Atropine
Atropine may be administered in the setting of bradycardia,5 although it may not always be effective in increasing heart rate in severely poisoned patients and may even worsen bradycardia in case of conduction disturbances predominantly located distally to the AV node (table 1 and 2).
Glucagon
Glucagon stimulates adenyl cyclase, increasing intracellular cyclic adenosine monophosphate and calcium influx into myocytes.2 The clinical efficacy of glucagon as an antidote for CCA toxicity is uncertain. In animal models of CCA poisoning, glucagon generally improved heart rate and cardiac output, but human data show variable results.28,39 Therefore, glucagon may not be the most suitable treatment option for CCA poisoning. However, in case of combined CCA and beta-blocker intoxication, glucagon should be considered.
Intravenous lipid emulsion
Intravenous lipid emulsion (ILE) is a relatively new intervention, used in the treatment of cardiovascular instability unresponsive to supportive care during intoxication with lipophilic drugs.40-45
The proposed mechanism of action is intravascular sequestration of the toxic drug to a newly formed intravascular lipid phase (‘lipid sink’), decreasing the free blood concentration of the lipophilic drug. This in turn leads to redistribution of the lipophilic drug from the target tissues into the blood, lowering drug concentration in the target tissues.46 Alternatively, ILE might serve as energy source for cardiomyocytes.46 Moreover, ILE could induce inotropic effects,47 likely via increasing intracellular calcium levels in cardiomyocytes.
Several case reports show beneficial effects of ILE, although ILE was not successful in all CCA-overdosed patients.28 Nevertheless, ILE should be considered in patients with life-threatening cardiovascular toxicity, who show an insufficient response to aforementioned conventional therapies. Notably, ILE has been used as parenteral nutrition for many years, with an acceptable safety profile, and is available in most hospitals.
Extracorporeal life support
When supportive and pharmacological interventions are not sufficiently effective, extracorporeal life support (ECLS) should be considered.48 ECLS was shown to increase survival in patients with severe shock or cardiac arrest following poisoning due to drug overdose.28,48,49 Due to potential adverse events of ECLS, i.e. thromboembolism, haemorrhage, infection and limb ischaemia, ECLS should only be applied in experienced centres.
Other therapies
The use of a pacemaker should be considered when pharmacological interventions fail to improve haemodynamically significant bradycardia. Treatment options also proposed for CCA overdose are phosphodiesterase III inhibitors, levosimendan, L-carnitine, methylene blue and intra-aortic balloon pump.1,2,28 Information on the efficacy and safety of these options is sparse. Further studies should be conducted before any of these therapies are implemented in CCA overdose.
SUMMARY AND CONCLUSION
CCA overdose can cause life-threatening intoxications with predominant cardiovascular instability. Patients in whom a moderate to severe intoxication is anticipated should be observed in a high-care setting and treatment should first be aimed at gastrointestinal decontamination and supportive care, i.e. the administration of intravenous fluids, and correction of metabolic acidosis and electrolytes. Moderately/severely intoxicated patients may require a well-tailored combination of interventions, e.g. the administration of calcium, high-dose insulin-glucose therapy and vasopressors. Especially hyperinsulinaemia/ euglycaemia therapy is promising for patients in whom a large ingestion of a CCA is suspected, which should be initiated early in the stage of intoxication even when the patient shows hardly any haemodynamic instability. ILE may be considered in patients with life-threatening cardiovascular toxicity, with an insufficient response to conventional therapies. If clinical stabilisation cannot be achieved after applying conventional pharmacological measures, the initiation of ECLS should be considered. The efficacy and safety of the treatments currently available for CCA poisoning, including optimal dosing strategies, should be further explored in controlled clinical trials, which would optimise evidence-based decision making in managing CCA-overdosed patients.
DISCLOSURES
The authors declare no conflict of interest. No funding or financial support was received.
REFERENCES