
KEYWORDS
AA amyloidosis, hyperimmunoglobulinaemia D and periodic fever syndrome (HIDS), mevalonate kinase deficiency (MKD), anakinra; IL-1 blockage.
INTRODUCTION
Hyperimmunoglobulinaemia D and periodic fever syndrome (HIDS), a hereditary periodic fever syndrome, is an autosomal-recessive, auto-inflammatory disorder mediated by mevalonate kinase deficiency. The main symptoms are recurrent febrile attacks, arthralgia, cervical lymphadenopathy, diarrhoea and rash.1 Diagnosis is confirmed by genetic analysis since multiple mutations have been described in the mevalonate kinase (MVK) gene.2 Little is known about the mechanism leading to an auto-inflammatory condition as a result of reduced MVK activity. Ex-vivo experiments suggest that peripheral blood mononuclear cells produce large amounts of IL-1, hypothetically as a result of either excess or lack of isoprenoid products.3 Recently, it was hypothesised that a lack of 25-hydroxycholesterol, a metabolite in the cholesterol pathway, plays a role in the pathogenesis.4
AA amyloidosis is a known complication of hereditary periodic fever syndromes, especially familial Mediterranean fever in which the incidence has been reported to be as high as 60-75%.5 Reactive amyloidosis secondary to HIDS is relatively rare. Only six cases of amyloidosis secondary to HIDS have been reported in the literature.6-10 Literature on the treatment of patients with AA amyloidosis secondary to hereditary periodic fever syndromes other than familial Mediterranean fever is very sparse.
We present the seventh case of a patient with reactive AA amyloidosis secondary to HIDS and evaluate the response to IL-1 blockage in the progression of amyloidosis.
CASE REPORT
We present a 62-year-old female patient with genetically confirmed HIDS. She was heterozygote for the I268T and V377I mutations of the MKV gene. The patient was admitted with chronic diarrhoea. During admission she developed a rapidly progressive nephrotic syndrome. Physical examination revealed marked oedema up to the patient’s waist. Laboratory results showed: serum creatinine levels up to 194 µmol/l corresponding to an estimated glomerular filtration rate of 23 ml/min/1.73 m2 , blood urea nitrogen 10 mmol/l, phosphate 1.73 mmol/l, albumin 20 g/l and cholesterol 6.8 mmol/l. The highest concentration of serum amyloid A was 15 mg/l (reference value 0-4 mg/l). Mevalonic acid in the urine had a concentration of 3 μmol/mmol (reference value < 1 μmol/ mmol).
Colonic and renal histological biopsies showed deposition of amyloid, present in small arteries and in the kidney, also in glomeruli. Positive Congo-red staining is shown in figure 1. Immunohistochemical staining for AA amyloid (antibody clone MC1, Dako bv, Belgium) was positive (figure 1).
Echocardiography, performed to assess cardiac involvement, revealed concentric hypertrophy with an echographic aspect of cardiac amyloidosis. On the computed tomography scan of the abdomen the walls of the terminal ileum and sigmoid showed signal enhancement and wall thickening.
Initial treatment with high-dosed corticosteroids showed no improvement in the patient’s intestinal symptoms or proteinuria, hence treatment with anakinra, an IL-1 receptor antagonist (100 mg/day subcutaneously) was initiated. Several months after initiation of therapy with anakinra, the proteinuria persisted and no improvement in renal function was observed; for this reason the line of treatment was discontinued. However, after cessation of IL-1 blockage, a marked exacerbation of the intestinal symptoms was noted. Gastric biopsy revealed amyloid depositions in the gastric microvasculature. Nine months after the initial diagnosis of reactive amyloidosis without any amelioration of the symptoms and a decreasing quality of life our patient declined further treatment and died soon after.
DISCUSSION
We present the seventh case of AA amyloidosis complicating HIDS. Amyloidosis is often suspected in hereditary periodic fever syndromes in particular familial Mediterranean fever, cryopyrin-associated periodic syndromes (CAPS; e.g. Muckle-Wells syndrome) and tumour necrosis factor receptor-associated periodic syndrome (TRAPS). However, in HIDS this major complication is rarely seen despite the favourable conditions for amyloidogenesis.11 The defect in mevalonate kinase, which leads to a deficiency of isoprenoid products, seems to have a protective effect on the development of amyloidosis. Farnesylated proteins seem to play an important role here.12 Early diagnosis is crucial because limiting the progression of amyloidosis by treating the underlying disease is the cornerstone of the management strategy.
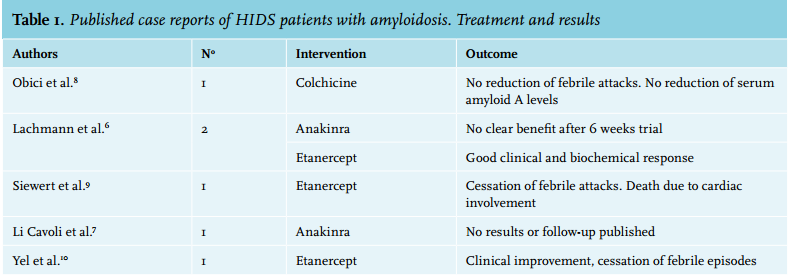
No directed therapy for HIDS is currently available. Recently a comprehensive review of the treatments of hereditary periodic fever syndromes was published by Ter Haar et al.13 Specific data on the treatment of HIDS in this review suggest that colchicine is not effective.13-15 Statins were a promising therapy due their blocking effect in the isoprenoid pathway but results were also poor.13-15 NSAIDs and corticosteroids are commonly prescribed in clinical practice and are anecdotally reported to have positive results in HIDS.14,15 Therapy with anakinra, an IL-1 inhibitor and etanercept, a TNF-α inhibitor, has shown overall positive results.13-22 Recent publications with canakinumab, an IL-1 antibody, have also shown promising results.15,23 The question remains what the therapy of amyloidosis secondary to HIDS should be. In table 1 we summarise the results from previous case reports and small case series in the literature. Colchicine was found to be ineffective in one patient.8 Two reports of treatment with anakinra were published, the clinical course was not described in one of the reports and no clear benefit was seen in the other.6,10 Etanercept was reported to have good results in three patients.6,7,9
In the absence of a clear evidence-based treatment strategy we propose following a step-up plan for future cases. Stopping amyloidogenesis must be achieved by blocking the acute-phase response. Guidelines recommend initiating steroids but experts doubt their effectiveness. If the amyloidosis aggravates, the next step should be one of the biological drugs discussed above. Expert consensus is that anakinra may be more effective than TNF blockade, however this is based mainly on pathophysiological arguments and clinical experience. Anakinra has been the most effective biological drug for HIDS but has not been as effective in cases complicated by amyloidosis. Higher dosages of anakinra might be needed and timing of initiation of therapy is of major importance. The choice of a specific biological cannot be deducted from the literature and should be tailored to each patient based on individual characteristics. The key to limiting amyloid deposition and its target organ complications is early recognition and diagnosis. 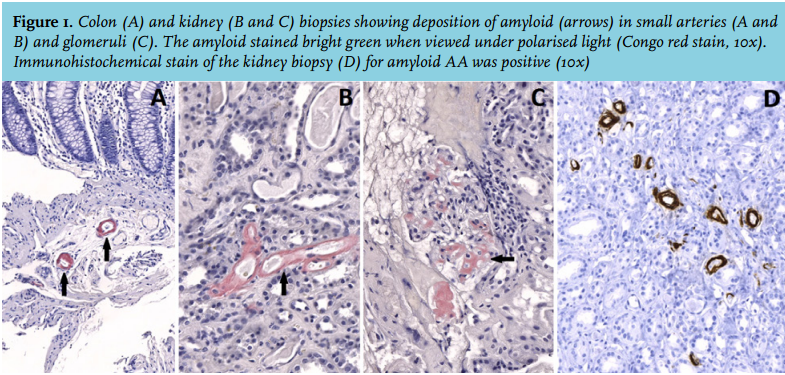
DISCLOSURES
The authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES