KEYWORDS
Acute intermittent porphyria, diagnosis, erythropoietic porphyria, porphyria cutanea tarda, porphyrias
INTRODUCTION
In the majority of patients, porphyria is caused by an inherited deficiency of an enzyme of haem biosynthesis and can be classified as acute or cutaneous or a combination of both. Acute porphyrias are characterised by attacks of acute abdominal pain, often accompanied by neurological symptoms. Hypertension, chronic kidney disease, and hepatocellular carcinoma are major long-term complications of the acute porphyrias. Cutaneous porphyrias are characterised by severe pain or blistering of the light-exposed areas of the skin.
Attacks in acute porphyrias only occur in a small percentage of gene carriers (< 10%) and are often precipitated by non-inherited factors, such as the ingestion of certain drugs, weight loss, or hormonal changes. Undiagnosed acute porphyria can lead to continuance of the precipitating factors and inadequate treatment, and result in progressive neurological problems including paralysis, seizures, and coma.1 Once a diagnosis has been made, the risk of attacks may be lowered by reducing exposure to precipitating factors; the severity of attacks may also be diminished by timely treatment.
Cutaneous porphyrias are rarely life-threatening, but can cause severe social isolation and a low quality-of-life due to avoidance of light exposure to prevent pain, or to reduce fragility or disfigurement of the skin.2,3
The aim of this review is to increase awareness of porphyrias and to prevent diagnostic and therapeutic delay. We provide basic information on porphyrias and practical flow charts for diagnosis and treatment to be used by health care professionals (e.g., general practitioners, internists, gastroenterologists, surgeons, neurologists, psychiatrists, and dermatologists).
Acute porphyrias
Acute porphyrias are characterised by acute attacks of severe pain, most frequently abdominal pain, sometimes only in the back or upper legs. This pain can be accompanied or preceded by symptoms such as anxiety, depression, irritability, and mood changes and followed by progressive neuropathy.4-8 There are four different types of porphyrias that can present with acute porphyric attacks: acute intermittent porphyria (AIP), variegate porphyria (VP), hereditary coproporphyria (HCP), and delta-aminolaevulinic acid dehydratase deficiency (ALAD).9 AIP is by far the most prevalent acute porphyria in Europe, whereas ALAD is extremely rare, with eight cases reported worldwide.10 VP and HCP are acute porphyrias that can also present with cutaneous blistering in sunlight-exposed skin. The four different types of acute porphyria are all caused by pathogenic variants in autosomal genes, resulting in reduced activity of one of the enzymes in the haem biosynthesis pathway (table 1). Although the affected genes and enzymes differ per type of acute porphyria, the pathophysiology of these attacks is similar in all types of acute porphyria. Other than inherited deficiencies in the haem synthetic pathway, there are other rare causes of acute porphyric attacks, such as lead intoxication and tyrosinemia; lead and succinyl acetone, respectively inhibit ALAS1. The first step to diagnosing an acute attack is the same in all causes of acute porphyria. In this review, AIP will be discussed in detail. 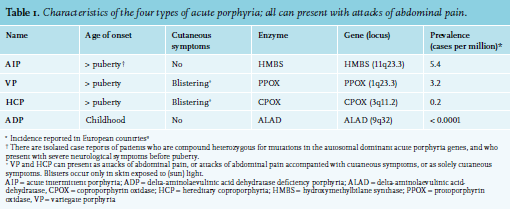
Pathophysiology of acute attacks
Porphyria symptoms result from the accumulation of intermediates of haem biosynthesis. Acute porphyric attacks are related to an increase in ALA levels11 and ALA accumulation seems to be the main culprit in acute porphyric attacks. The exact mechanism whereby ALA causes the neurological symptoms is unknown. Acute porphyric attacks are seen in conditions with isolated increases in ALA without a rise in porphobilinogen (PBG), including tyrosinemia, lead poisoning, and ALAD deficiency. Attacks can be provoked by precipitating factors that induce ALAS1 and ALA levels, either directly or via haem depletion. Well-described provoking factors are cytochrome P450-inducing drugs, infections, fasting, and alcohol use.12-16 Haem production by hepatocytes is regulated differently to that in erythroid cells. In the liver, haem as an end-product, regulates haem production (figure 1) by suppressing ALAS1 expression at the transcriptional, translational, and post-translational levels.17,18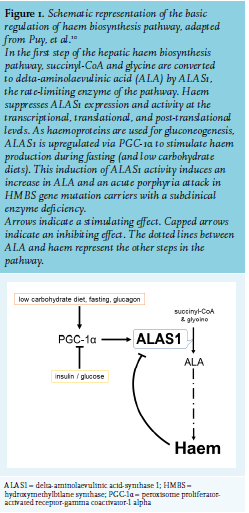
Symptoms, demographics, and complications of acute porphyrias
In all forms of acute porphyria, an acute attack may start with changes in behaviour and restlessness. During an attack, nearly all patients have severe abdominal pain. This can be accompanied by anxiety, vomiting, constipation, diarrhoea, tachycardia, hypertension, hyponatraemia, red urine, muscle weakness, and neurological symptoms such as paraesthesia, paralysis, and seizures. Severe motor neuropathy can lead to respiratory failure and posterior reversible encephalopathy syndrome, with headaches, visual symptoms, and seizures, which have been observed during attacks. If left untreated, severe neurological damage and even death, can occur. At the beginning of an attack, patients suffer from anxiety and pain, without signs of obstruction or peritonitis on physical examination. The lack of physical signs may result in diagnoses of neurasthenia or hysteria, which can be emotionally traumatic.
The majority of individuals with pathogenic hydroxymethylbilane synthase (HMBS) variants remain asymptomatic, as only a minority (1-10%) of all HMBS gene mutation carriers experience an acute attack during their lifetime.19 Generally, symptomatic patients do not present before puberty and most patients are females of the reproductive age; however, acute porphyric attacks can occur in both men and women.5,10 Although most symptomatic patients have only one or two attacks during their lives, a small subset of the symptomatic patients suffer from recurring attacks (≥ 4 hospitalisations due to attacks per year).20,21 Both symptomatic patients and asymptomatic gene mutation carriers of any of the acute porphyrias have an increased lifelong risk of hypertension, kidney disease, and hepatocellular carcinoma (HCC), but the incidence differs between porphyria types and countries.22-23
What should be investigated?
When an acute porphyric attack is considered, urinary ALA, preferably together with PBG, should be measured. This can be performed in a urine sample; 24-hour collections are not necessary.24 An acute porphyric attack can be considered when ALA levels (or, if an ALA assay is unavailable, PBG levels) are increased to at least twice the upper limit of normal (ULN). Diagnostic guidelines are in progress in Europe and the United States (US), and there is debate on what the cut-off value should be; twice ULN can be considered conservative and safe regarding false-negative interpretations. Importantly, the urine sample should be protected from light (using aluminium foil) and kept refrigerated (< 4 ºC) while being sent to the laboratory. Protecting a sample from light during transport is necessary to prevent false-negative findings, since light stimulates non-enzymatic isomerisation and changing of ALA/PBG into porphyrins and other metabolites. After attacks, urinary ALA and PBG levels can return to baseline levels, but in most AIP patients this normalisation takes years.25 Plasma ALA and PBG levels are of pivotal importance in patients that are anuric. Plasma ALA/PBG levels can remain elevated much longer than urinary levels after an attack subsides and as such, might be an important biological marker in patients that present asymptomatically but have had suspected attacks in the past.26-27
Mild ALA increases (twice ULN) can also be explained by certain drugs, heavy metals such as lead, renal disease, and liver disease.28 After confirming an acute porphyric attack, treatment should be started without delay. The type of porphyria does not affect the initial treatment, however, following initiation of treatment, further diagnostic steps should determine the specific type of acute porphyria (AIP, VP, HCP, ALAD). For rare cases with acquired porphyria such as lead intoxication, additional steps such as preventing the patient’s exposure to the toxicant are essential to cure the patient. There are porphyria expert centres in most European countries, where the required diagnostic tests and the interpretation can be performed.29 The ALA/PBG ratio, HMBS enzyme activity in erythrocytes, and plasma (or faeces) porphyrin spectrum can guide clinicians to the correct porphyria type. DNA gene mutation analysis can be used as a confirmation step. If a DNA gene mutation cannot be detected, additional enzyme activity can be measured. The specific DNA gene mutation can be used for family counselling. If no mutation in the coding region can be found, a diagnosis can be confirmed by measuring the appropriate enzyme activity (e.g., erythrocyte/lymphocyte porphobilinogen deaminase activity in AIP).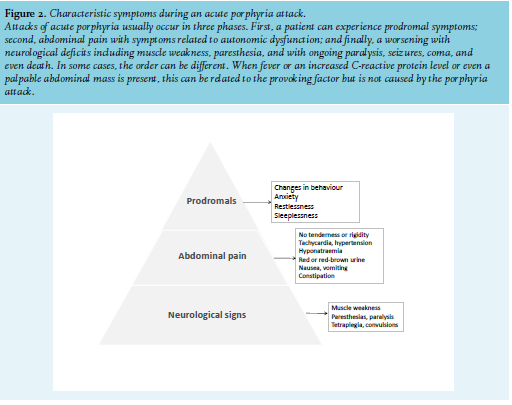
Treatment of an acute porphyric attack 30,31
Treatment should be started as soon as possible in known acute porphyria patients presenting with severe symptoms, like neurological symptoms, or hyponatraemia, even before the results of confirmatory tests are performed. Treatment should also be started in severe cases without waiting for confirmatory ALA/PBG tests (table 2). In milder cases, without neurological symptoms, it is recommended to start with oral (preferable), and otherwise parental, carbohydrate loading; for example, 200 g or the equivalent, such as 2 litres 10% glucose per day (except in patients with hyponatraemia; they should only receive carbohydrate via the enteral route and haem should be considered immediately). It is also very important to remove any provoking factor(s). Clinical evidence on the effectiveness of a high doses of carbohydrates in suppressing attacks is limited, but there is a good theoretical basis for providing extra carbohydrates as they have demonstrated inhibition of haem synthesis in hepatocytes via PGC-1a (peroxisome proliferator-activated receptor-γ coactivator-1a); carbohydrate-restricted diets are notorious for provoking attacks. During an attack, patients are at risk of developing hyponatraemia and plasma sodium should be initially measured every three hours. Hyponatraemia should be evaluated and treated according to hyponatraemia guidelines. The exact pathogenesis of hyponatraemia in acute porphyrias is not clear; in most cases, findings point towards an inappropriate secretion of antidiuretic hormone and therefore, high-dose glucose infusion should be stopped immediately. Pain control during attacks can be difficult to achieve and high-dose opioids are often needed. In severe attacks and those with significant hyponatraemia, haem arginate should be given immediately. Haem arginate must be dissolved in 20% albumin to prevent phlebitis and given at a dose of 3 mg/ kg daily for three to four successive days (one ampoule of haem arginate (Normosang®) contains 250 mg which is sufficient for most adults). The bottle and tubes containing haem need to be protected against light (by aluminium foil wrapping or other). The presence of hyponatraemia, neurological signs, and/or severe non-responsive pain are markers of a severe attack and such patients should be admitted to an intensive care unit.
Haem administration leads to a rapid decrease in ALA and PBG over a period of days, and sufficient pain reduction can take additional time. Peripheral neuropathy and paralysis take weeks to months to improve, but haem therapy can prevent progression to paralysis, seizures, coma, and death.32
Hypertension can be exacerbated by the pain, anxiety, and stress, but is also considered to be caused by autonomic dysfunction due to the neurotoxicity of ALA. Beta-blockers, such as propranolol, are safe and effective for reducing blood pressure and tachycardia. All medication, either prescribed or over the counter, should be cross referenced for safety with the Norwegian Porphyria Centre Drug Database for Acute Porphyria (www.drugs-porphyria.org). In this database, alternative drug options are provided per indication. Drugs that are not safe or possibly harmful, including herbal medications (e.g., St John’s wort), should also be discontinued. 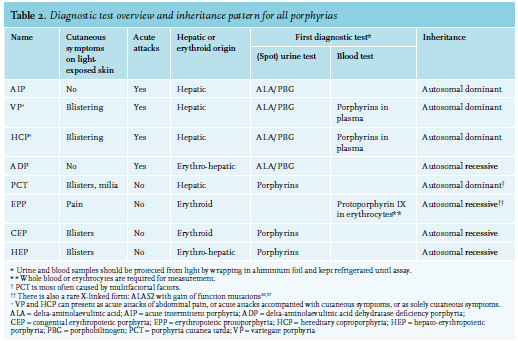
Follow-up of symptomatic and asymptomatic patients
In general, patients should be counselled about their disease and informed about lowering their risk of attacks by avoiding provoking factors, including drugs, alcohol, and smoking. All new prescriptions should be checked regarding safety.33 Surgical procedures warrant pre-operative planning and, in circumstances with risk of low carbohydrate intake, measures should be discussed. Each patient should have an emergency plan and a medic alert, with what to do and monitor in these situations.34 Blood pressure and renal function should be monitored lifelong on a regular basis. Deterioration of kidney function in AIP patients is independent of hypertension, suggesting a secondary mechanism for kidney damage.35 One proposed mechanism is the direct toxic effect of ALA, causing tubular atrophy, interstitial fibrosis, and chronic arteriolopathy.36
An increased risk for hepatocellular carcinoma (HCC) in acute porphyria patients is well recognised and is unrelated to liver fibrosis or cirrhosis.37-39 The pathophysiological mechanism has not been elucidated.40 Considering this risk, it is advised to screen patients over 50 years of age with a bi-annual liver ultrasound, similar to the HCC screening schedule in hepatitis-induced liver cirrhosis patients. All first-degree relatives should be counselled by a clinical geneticist in a porphyria expert centre,29 considering their 50% chance of having the same HMBS gene mutation.
Patients with recurrent attacks
A small subset of patients has frequent attacks (≥ 4 per year) that often require hospitalisation. These patients report significantly more porphyria-related symptoms, also in between attacks. They have more chronic complications and a lower quality of life.7,8 Current treatment options include prophylaxis by weekly haem infusion.41
A promising emerging therapy for patients with recurrent attacks is the suppression of ALAS1 activity by administering small interfering RNA (siRNA), givosiran (Alnylam Pharmaceuticals, Cambridge, USA). A phase III trial (ClinicalTrials.gov – NCT03338816) shows impressive and significant reductions in attack frequencies and urinary ALA (Envision, A phase 3 study of safety and efficacy of givorisan, an investigational RNAi therapeutic, in acute hepatic porphyria patients. Gouya L & Sardh E. International Congress on Porphyins and Porphyrias 2019, Milano, Italy). In November 2019, givosiran was approved by the US Food and Drug Administration and in March 2020, by the European Commission/European Medicines Agency (EMA).42,43 Further studies on the long-term effects and safety of this drug are ongoing; an international phase III trial demonstrated a significant reduction in the number of attacks and levels of ALA and PBG81 A liver transplantation is currently the only available curative treatment for AIP. It replaces the deficient hepatic enzyme and prevents future attacks, but liver transplantation is not always possible and remains a high-risk procedure.44-48 Another emerging therapy still in the pre-clinical phase is messenger RNA for AIP patients (mRNA, Moderna Ltd, Cambridge, USA).49
Cutaneous porphyrias
The cutaneous porphyrias are porphyria cutanea tarda (PCT); erythropoietic protoporphyria (EPP); and four rare forms, including hereditary coproporphyria, variegate porphyria, and extremely rare, hepato-erythropoietic porphyria (HEP) and congenital erythropoietic porphyria (CEP) (table 3).50 The symptoms of cutaneous porphyrias include blistering with scarring, milia, hypo-depigmentation, skin fragility, photomutilation, and very painful phototoxicity.2,51 Figure 3 provides a diagnostic flowchart for distinguishing the main cutaneous porphyrias.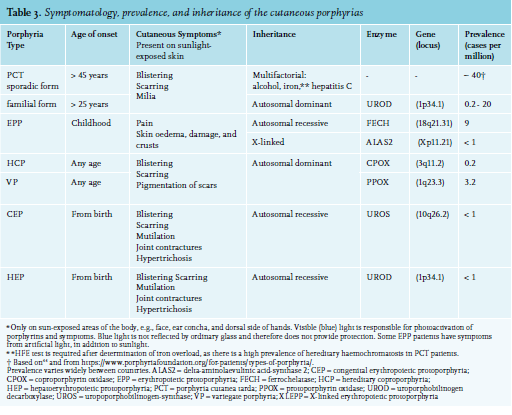
Pathophysiology of cutaneous porphyrias
Porphyrin molecules are formed or modified during the last six steps of haem biosynthesis. Porphyrin molecules are excited by visible light, especially blue light, and to a much lesser degree, UVA light of near violet wavelengths. The porphyrins accumulate in the skin and/or erythrocytes, and exposure to light leads to phototoxic damage by energy released from the porphyrins. The energy released from porphyrins results in the formation of oxygen radicals and causes local haemolysis, histamine release from mast cells, and endothelial damage resulting in tissue damage. In contrast to UV-light, blue light is not absorbed by glass and therefore patients are not protected by glass or by UV sunblocks or sunscreens, which mainly block UV-B light. Patients can only shield themselves from blue light by avoiding exposure and staying indoors, or covering their skin by wearing protective clothing such as long sleeves, long pants, socks, gloves, and broad-brimmed hats. PCT and EPP will be discussed in more detail.
Porphyria cutanea tarda
In PCT, accumulation of the photo-reactive uroporphyrin III in the epidermis causes painless blisters in sunlightexposed areas of the skin and an increased fragility of the skin. PCT is caused by decreased activity of the hepatic enzyme uroporphyrinogen-decarboxylase (UROD). This enzyme catalyses the conversion of uroporphyrinogen to coproporphyrinogen. PCT occurs as hereditary or sporadic forms. In sporadic PCT, external provoking factors include alcohol consumption, hereditary haemochromatosis, hepatitis C, smoking, oestrogens, renal insufficiency, exposure to certain chemicals, or a combination of these. The reported association between human immunodeficiency virus (HIV) and PCT is almost certainly due to co-infection with hepatitis C.52 In hereditary PCT, patients have a pathogenic gene mutation in the UROD gene which causes decreased UROD activity. Only a small percentage of UROD gene mutation carriers will develop symptoms, and external factors are still required to increase uroporphyrin levels and develop symptoms.53 Hereditary PCT accounts for approximately 25-50% of all PCT cases, with large differences in prevalence between different countries.54 Hereditary PCT patients present at a younger age, in the third or fourth decade of life, while patients with the sporadic form usually present in their fifties or later. In both forms, there is an almost invariable association with some degree of iron overload.
When to suspect PCT?
PCT should be suspected in patients with painless blisters and lesions on the back of the hands and/or face and the ears, and in late onset hirsutism. The skin becomes fragile and minor trauma or rubbing can easily peel skin off, causing open lesions, crust formation, and eventually scarring and milia. Blisters and scars in skin not exposed to sunlight are very unlikely to be due to PCT.
What should be investigated?
A porphyrin spectrum in a spot urine sample should be tested. Patients with PCT have a specific pattern of porphyrins, with increased levels of uro-, hepta-, and hexaporphyrin (table 2). The urine sample must be protected from light (packed in aluminium foil) and kept refrigerated (< 4 °C) until analysis.
The following tests should be performed to define the provoking factors: renal function, liver enzymes, iron status, hepatitis C, and HIV serology. When raised serum ferritin levels are associated with increased transferrin saturation, diagnostics for hereditary haemochromatosis (HFE gene sequencing) should be performed. In rare cases, high dose oestrogens, haematological malignancy, or liver adenomas can provoke PCT. As there is an increased incidence of liver tumours in PCT, a liver ultrasound is recommended at least once, and more frequently if other risk factors for hepatocellular carcinoma such as cirrhosis are present.55
Treatment and follow-up of PCT
Patients with active PCT should stop drinking alcohol, stop smoking, and avoid direct sunlight completely until symptoms resolve and biochemistry is normalised. Patients should consider stopping oestrogen use. Treatment of PCT is nearly always successful. The treatment consists of eliminating provoking factors and reducing iron stores by phlebotomies or chelation in all patients. Phlebotomies should be continued until urinary uroporphyrin levels are near normal and ferritin is at least < 50 µg/l, whilst maintaining normal haemoglobin levels; this recommendation is based on the Dutch hemochromatosis guideline.56
When phlebotomies are contraindicated or not tolerated, hydroxychloroquine can be considered in a low dose (start with 200 mg once a week or 100 mg twice a week).50 In hepatic lysosomes, hydroxychloroquine forms soluble complexes with the accumulated porphyrins (uroporphyrin) and subsequently increases urinary porphyrin excretion, resulting in a darkening of the urine.57,58 In addition to increasing porphyrin excretion, it has been suggested that chloroquine and hydroxychloroquine increase urinary iron excretion.59 High doses of chloroquine and hydroxychloroquine, such as given in rheumatology, must be avoided in PCT as they result in hepatotoxicity.60 Whether ophthalmological screening is necessary is doubtful as retinopathy has never been described with the low dose and the treatment duration is often less than one year.61,62 According to the guidelines of the Dutch Society of Rheumatologists, ophthalmological screening should be considered in PCT patients with other risk factors such as renal impairment, concomitant treatment with tamoxifen, or the presence of maculopathy or retinopathy.63 Most patients achieve biochemical remission within nine months.57 Some patients continue to experience skin fragility after biochemical remission.
Erythropoietic protoporphyria (EPP)
Erythropoietic protoporphyria patients are intolerant to light. The photosensitivity usually starts in early childhood and is lifelong. EPP is caused by ferrochelatase deficiency (FECH) or by increased activity of the erythroid specific delta-aminolaevulinic acid synthase 2 (ALAS2). In 1-2% of EPP patients, the disorder is caused by a gain of function variant in ALAS2.64,65 EPP patients accumulate protoporphyrin IX (PPIX) in their erythrocytes. Signs in the skin of EPP patients, such as erythema and oedema may not be immediately visible to the physician when the patient presents with severe pain. The pain and skin damage in EPP patients, is deeper in the skin compared to damage in the skin of a PCT patient. Light exposure of protoporphyrin IX (PPIX) in erythrocytes circulating in small skin vessels results in an energy release with oxygen radical formation, causing endothelial and skin damage. This process results in extreme pain and can occur after a few minutes up to 30 minutes in sunlight. The pain can last for days after withdrawal from light, and may be followed by erythema, oedema, and petechiae. Blistering is uncommon. The differences in skin changes between EPP and PCT (and HCP and VP) are thought to be due to the localisation of the porphyrins in the skin and possibly in the cell organelles. In EPP, the initial site of damage seems to be in the endothelium and in PCT in the interstitium.
When to suspect EPP?
EPP should be suspected in children and adults who present with severe pain and/or oedema in reaction to light exposure. The majority of patients experience their first symptoms during early childhood. At a very young age, they can present with excessive crying. Children often learn to avoid light even before the diagnosis is made. Most patients describe a prodromal phase, shortly after exposure to (sun)light. This is described as a burning, tingling, or itching sensation in the skin. When light exposure is prolonged, pain will increase, and eventually oedema, erythema, petechiae, necrosis, and crusts may be seen. The pain does not respond to analgesics, including opioids, and can last up to a week. These painful episodes result in light-avoiding behaviour from an early age, with a diminished quality of life, anxiety, vitamin D deficiency, and a negative impact on social life. The amount of time patients can stay in direct sunlight is highly variable between patients and is influenced by weather conditions. Windy, cold sunny days are the worst. EPP symptoms can occur during light exposure behind glass, as blue light is not filtered by glass.
What should be investigated?
To diagnose EPP, free erythrocyte PPIX levels should be elevated by > 3 times the ULN (table 2). The blood sample must be shielded from direct light (packed in aluminium foil) and kept refrigerated but not frozen (4°C). Both free protoporphyrin and zinc protoporphyrin are measured. In addition to protoporphyrin levels, zinc protoporphyrin levels are markedly raised in ALAS2 X-linked protoporphyria, and the ratio can help differentiate between ALAS2- and FECH-related EPP. Zinc protoporphyrin levels are also increased in iron deficiency and lead poisoning. Most patients will have a loss of function FECH mutation combined with a common polymorphism, which causes mis-splicing and reduced expression of normal mRNA.64 A diagnosis can be genetically confirmed with FECH or ALAS2 gene mutation analyses (table 2).
Treatment and follow-up of EPP
Until 2016, EPP patients could only avoid light exposure as there was no effective treatment registered for EPP except bone marrow transplantation, which cures EPP but has great health risks. UV-B therapy, beta-carotene, and cysteine have been tried, but effects were unsatisfactory.67 In 2014, the EMA approved afamelanotide (a synthetic alpha-melanocyte stimulating hormone, Scenesse®, Clinuvel Pharmaceuticals Limited, Melbourne, Australia) marketing authorisation for adult EPP patients in the European Union and in 2016, treatment became available and reimbursed in the Netherlands. Afamelanotide binds to the melanocortin 1 receptor thereby increasing the production of photoprotective eumelanin in the skin. Patients who are currently treated with afamelanotide (subcutaneous implant with 16 mg, 1-4 times per year) report an increase in symptom-free sunlight exposure time, a decrease of the phototoxic effect, and an improved quality of life with only mild adverse events (nausea, headaches, flu-like symptoms, or flushes).68-69 Afamelanotide treatment can only be given in specialised porphyria expert centres, and is currently available in Switzerland, Italy, Germany, Austria, and the Netherlands. In the remaining European countries, reimbursement issues are still a problem. In the US, the FDA approved afamelanotide in October 2019.70
All patients with EPP should be screened for vitamin D deficiency and nearly all require vitamin D supplementation. Osteoporosis and osteopenia are more prevalent in EPP patients and occur at a younger age compared to the general population.71 Dual-energy X-ray scans are recommended at least once in adult patients.
Approximately 5-20% of patients with EPP develop liver disease.72 In plasma, free PPIX binds to albumin and is taken up by the liver and excreted via the bile ducts into the faeces. PPIX damages bile duct cells and the hepatic parenchyma. Patients with EPP have an increased risk of gallstones (pigment stones containing protoporphyrin). An EPP-related acute cholestatic hepatitis is the most serious complication of EPP, occurring in fewer than 5% of the patients.73-75 During a hepatic crisis, PPIX excretion is decreased, resulting in a steep rise of blood and hepatic PPIX levels, causing more liver damage, with a vicious circle leading to more liver damage, haemolysis, and additionally a further rise in PPIX. The treatment of an EPP-related hepatitis should be in close collaboration with an expert centre. One treatment aim is to rapidly lower PPIX levels to stop additional liver damage and improve hepatic recovery. To rapidly lower PPIX levels, erythrocyte exchange transfusions in combination with additional blood transfusions are the best option. Haemoglobin levels should be maintained at levels that suppress endogenous erythropoiesis (preventing the production of PPIX-rich erythrocytes). Liver transplantation can be considered when this therapy fails. As PPIX is produced mainly by the bone marrow and because hepatopathy may recur, a subsequent bone marrow transplantation should be considered.75-76 An acute cholestatic hepatitis can be precipitated by other causes of liver disease, including viral hepatitis, hepatotoxic drugs, and excessive alcohol intake.77 As a preventive measure, all EPP patients are urged to refrain from alcohol and hepatotoxic medication, and immunisation against hepatitis A and B is advised.
Caution should be taken for EPP patients requiring surgery, since prolonged exposure (> 3 hours) to bright surgical lights can induce phototoxic tissue injury, which can be prevented by using yellow filters that block blue light.78-80
CONCLUSIONS
Awareness of the porphyrias is of great clinical importance as they are a diverse group of conditions with severe consequences and complications. When these diseases remain undiagnosed, serious and irreversible damage can occur. This can often be prevented, and early recognition and treatment of acute porphyric attacks can save lives. Simple flowcharts are provided to help diagnose porphyria or exclude porphyria from the differential diagnosis (figure 3). Management, treatment, and follow-up of porphyria patients should preferably be performed by or in cooperation with expert centres, and by physicians with experience in treating these disorders (www.porphyria.eu).
Take home messages
Acute porphyria
Cutaneous porphyrias
ACKNOWLEDGEMENTS
We recommend the EPNET websites with disease information for physicians and patients (www.porphyria. eu), and the Drug Database for Acute Porphyria (www. drugs-porphyria.org). The INVEST website (www.investof.nl) provides an emergency protocol for an acute porphyric attack. There are two patient organizations in the Netherlands for patients with porphyrias: www.epp.info (for patients with EPP) and www.pvap.nl (for patients with acute porphyrias). We would like to thank Paul Wilson for his input on the manuscript.
DISCLOSURES
J.G. Langendonk is involved in industry-sponsored trials initiated by Clinuvel Pharmaceuticals LTD, producer of Scenesse®; and Alnylam® Pharmaceuticals, the producer of Givosiran®. R.A. Neeleman’s position is supported by a grant from SLO (Stichting Lever Onderzoek), an Erasmus MC Foundation for Liver and Gastrointestinal Research.
REFERENCES