KEYWORDS
Autoinflammatory disease, Familial Mediterranean Fever, Tel-Hashomer diagnostic criteria
INTRODUCTION 
Familial Mediterranean Fever (FMF) is the earliest described and most prevalent hereditary autoinflammatory disease, first described in 1945.1,2 It is usually inherited as an autosomal recessive trait, but rare cases of dominant transmission have been described in patients with the M694V mutation. The FMF gene, MEFV, is located on chromosome 16p13.3, coding for the protein pyrin.3,4 Its clinical presentation may be infrequent in non-Mediterranean countries, leading to delay in diagnosis and treatment. Characteristically, patients suffer from recurrent self-limiting attacks of fever and pain caused by pleuritis, peritonitis, arthritis, or erysipelas-like skin lesions.5,6 The clinical diagnostic criteria are derived from a study at the Tel-Hashomer Medical Centre in Israel.7 The diagnosis of FMF should be considered when a patient of Mediterranean origin meets two major criteria, or one major and two minor criteria (table 1).
As the name suggests, FMF is most prevalent in populations originating from the Mediterranean region: Sephardic Jews, Armenians, Arabs, and Turks.5, 8-14 In these populations, the prevalence is estimated at around 100-400 per 100,000 inhabitants. Due to extensive immigration in the 20th century, FMF is also reported in non-Mediterranean regions. For example, Germany, Italia, Czech Republic, and Japan reported patients with this auto-inflammatory disorder.15-19 Except for few case reports, FMF is not well documented in the Netherlands.20-23 Approximately 800,000 migrants, originally coming from areas where FMF is endemic, are living in the Netherlands. The majority of them are living in cities such as Amsterdam.20
The objective of this retrospective study was to describe FMF patients in Amsterdam, focusing on clinical, demographic, and genetic characteristics in a population of different ancestries. A comparison was made with FMF patients living around both Mediterranean and non-Mediterranean regions based on previous reports.
MATERIALS AND METHODS
Study design
Medical records of 53 patients from 1990 until 2012 in a teaching hospital, Onze Lieve Vrouwe Gasthuis (OLVG)-West in Amsterdam, were retrospectively studied.
Case definition
The inclusion criteria were paediatric or adult patients who met the clinical diagnostic criteria from the Tel-Hashomer study.7 For each patient, the following clinical, demographic, and genetic characteristics were extracted from the medical record. Demographic characteristics: gender, age, country of origin, and consanguinity of parents.
Clinical characteristics: age at onset of symptoms and at diagnosis to calculate time from onset of symptoms to diagnosis; clinical features (fever, peritonitis, arthritis, pleuritis, and/or erysipelas-like skin lesions); laboratory findings during acute attacks (C-reactive protein and erythrocyte sedimentation rate); disease-related complications (proteinuria and/or amyloidosis; proteinuria was measured initially through urine dipstick screening, and 24-hour collection was performed in order to quantify proteinuria); abdominal surgery; response of treatment (measured in use of colchicine related to frequency of attacks before and after use); and disease severity score (table 2).24,25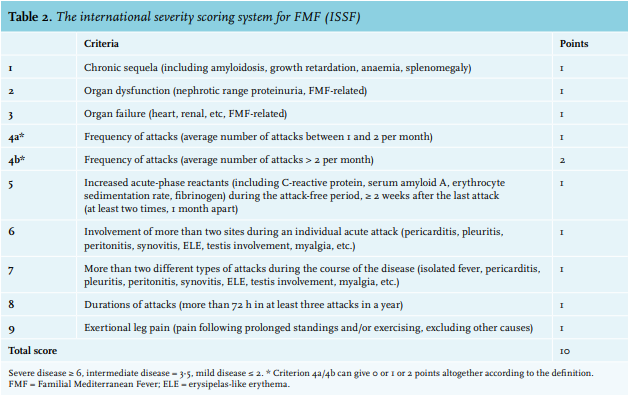
Genetic characteristics: genetic analysis was initially done by screening for known gene mutations in the MEFV gene. Since 2010, mutations in exon 1 until 10, inclusive of intron en exon transitions, were tested. Variants are described as homozygous, heterozygous, or compound heterozygous mutations.
Statistical analysis
Statistical analysis was performed using SPSS 21.0 (SPSS Inc., Chicago, IL). Results are expressed as means ± standard deviation (SD) for continuous variables and frequencies/ rates were measured for discrete variables. Means of the groups were compared with the Student t-test and one-way ANOVA test; a p-value < 0.05 was considered significant. The ethics committee of OLVG-West approved the study. An information letter instructed patients or their parents, in cases of minorities. All of them gave informed consent.
RESULTS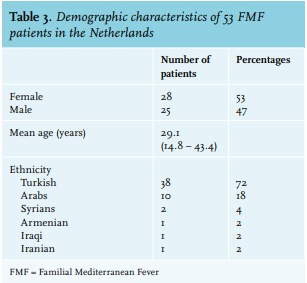
Demographic characteristics
Demographic characteristics of 53 FMF patients are demonstrated in table 3: 28 patients (53%) were female. Their mean age at the time of analysis was 29.1 years. The main ethnicity was Turkish (n = 38, 72%).
Clinical characteristics
Clinical characteristics are demonstrated in table 4. The mean age at onset of symptoms was 13.8 years. Twenty-three patients (43%) were younger than 10 years at the time of onset of first symptoms, 11 (21%) patients were between 10-19 years, 11 patients (21%) were between 20-29 years, 6 patients (11%) were between 30-39 years, and in 2 (4%) patients, this was unknown. The mean age at diagnosis was 22.0 years and the mean delay to diagnosis was 8.2 years. According to gender, mean age at onset of symptoms was significantly earlier in females (11.6 years) compared to males (16.0 years); p-value 0.000. Mean age at diagnosis was also significantly earlier in females (18.7 years) compared to males (25.1 years); p-value 0.000. However, there was no significant difference in mean time from onset of symptoms to diagnosis between both sexes: 7.1 and 9.1 years, respectively (p-value 0.420).
The main clinical features during acute attacks were abdominal pain reflecting peritonitis (91%) and fever (81%). Less frequent complaints were pain in the hip, knee, or ankle reflecting arthritis (34%), thoracic complaints reflecting pleuritis (34%), or erysipelas-like skin lesions (2%). Most patients had multiple (two or three) clinical features during acute attacks. Forty-nine patients (93%) experienced the same pattern of clinical features during recurrent attacks.
Inflammatory markers were elevated during attacks: the mean C-reactive protein value and erythrocyte sedimentation rate were 133 mg/l and 37 mm/first hour, respectively. One patient who was 20 years at onset of first symptoms and 26 years at diagnosis with a homozygote M694V mutation, had histologically-proven amyloidosis (kidney biopsy) with proteinuria (5.6 g/24h) as a disease-related complication. One other patient had proteinuria (5.4 g/24h) without histological examination. Seventeen (32%) patients underwent abdominal surgery, because there was a suspicion of appendicitis, cholecystitis, or adnexitis and the diagnosis of FMF was not considered. Pathological examination showed no evidence of infectious inflammation in all of these cases.
Most patients used colchicine 0.5mg once or twice a day (n = 15, 31% in both groups). In four patients (8%), no treatment was given, either because the attacks were infrequent (one patient) or because the patients refused treatment (three patients). Before treatment, most patients had multiple monthly attacks (n = 36, 75%). The use of colchicine reduced this frequency; for 28 patients (58%), the attack rate was reduced to several times a year, and for 11 patients (23%) to less than once a year. Nevertheless, nine patients (19%) were unresponsive to colchicine treatment.
Based on the internationally accepted disease severity score for FMF,23,24 most of the patients classified their disease as mild (n = 22, 42%). 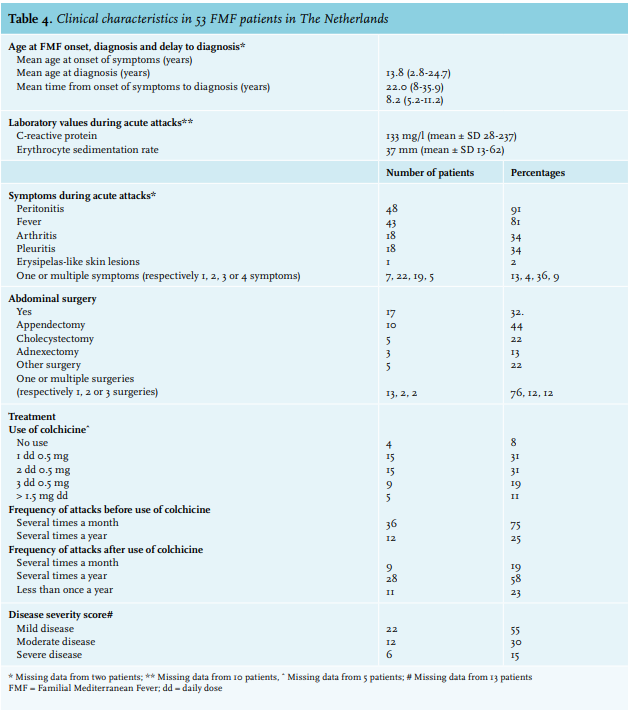
Genetic characteristics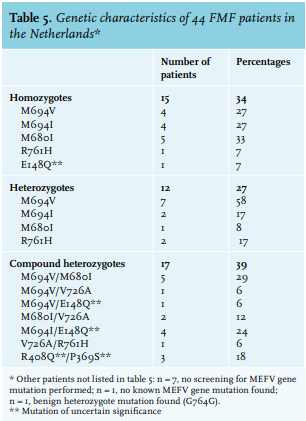
Screening of the MEFV gene for known gene mutations was performed in 46 patients and is demonstrated in table 5. In one patient, no known mutation in the MEFV gene was found and in one patient, a benign heterozygote mutation (G764G) was found. Most patients (n = 17, 39%) were compound heterozygotes, 15 patients (34%) were homozygotes and 12 patients (27%) were heterozygotes. The most frequent gene mutation was M694V which was found in 18 patients in total: in four patients as a homozygotes mutation, in seven patients as a heterozygotes mutation, and in seven patients as a compound heterozygotes mutation.
DISCUSSION
This retrospective study describes the first series of FMF patients in the Netherlands, focusing on clinical, demographic, and genetic characteristics. For early recognition and diagnosis of FMF in the Netherlands it is important to investigate and recognize the clinical presentation of FMF in non-Mediterranean European countries and compare these findings to the previously described spectrum in patients living in the Mediterranean region.
The main ethnicity in our population was Turkish, reflecting previous immigration from Turkey to the Netherlands, with a high estimated prevalence of FMF (1/1000) and high carrier frequency of 20% in the Turkish population.13 Some studies have reported a higher prevalence of FMF in males, others reported a similar female/male ratio, similar to our study population.9,12,14 All of our included patients have ancestors in endemic regions. Despite well-defined clinical diagnostic criteria by Tel-Hashomer, the episodic nature with short recurrent self-limiting attacks of fever and pain makes FMF a diagnostic challenge.5-8 Because of a low prevalence of FMF in non-Mediterranean regions, we expected a longer period from onset of symptoms to diagnosis in our study, because of the relative unfamiliarity of physicians with FMF. While symptoms of FMF started at a relatively older age than described in Mediterranean studies, we did not find a significantly longer delay of diagnosis. In our patients, symptoms started at a mean age of 13.8 years compared to 9.6 years in Turkish patients and to 67%, 80%, and 64% of the patients diagnosed with FMF before the age of 10 years in Jewish, Arab patients and Armenian patients, respectively.9,10,13 This finding is in contrast to other case series on FMF patients in non-Mediterranean countries.17,19 Our study showed similar delay from onset of symptoms to diagnosis compared to the Turkish FMF Study Group; 8.2 years compared to 6.9 years.13 Mediterranean studies reported delay of diagnosis ranging from 8 to 11 years.11,14 In other non-Mediterranean studies, there was a longer delay of diagnosis: 14.89 ± 10.10 years (median) in German patients, 18 years (9-27 years) in Italian patients and 9.1 years ± 9.3 years in Japanese patients.16,17,19
Clinical symptoms of Dutch FMF patients demonstrate a similar pattern as described in other Mediterranean and non-Mediterranean studies. Peritonitis (63-95%) and fever (78%-100%) are the most frequently described symptoms during acute attacks.9-11,13,15,17,19 While pleuritis was more frequently seen in non-Mediterranean studies compared to Mediterranean studies (49% vs 40%), arthritis and erysipelas-like skin lesions were more frequently seen in Mediterranean studies compared to non-Mediterranean studies (52% vs 32% and 22% vs 18%). Simliar to other studies, appendectomy and cholecystectomy were the most frequently performed abdominal operations (35% and 11% in Arabs patients and 19% and 2% in Turkish patients, compared to 44% and 23% in our study).11,13
Amyloidosis is the most serious complication of FMF, with a prevalence of 14% in non-Mediterranean studies and 13% in Mediterranean studies.9-11,13,15,17,19 We found a low frequency of amyloidosis: only one patient (2%) suffered from histologically-proven amyloidosis with proteinuria. Similar to previous genetic studies, the patient was homozygote for the M694V mutation, which is associated with a higher prevalence of amyloidosis.26 A possible explanation for this low incidence of amyloidosis in our study is the fact that colchicine was standard treatment for most FMF patients in our study.
In our study, 92% of the patients were treated with colchicine, which significantly reduced the frequency of attacks. Mediterranean studies reported a higher percentage of patients on colchicine and at a higher dose with better response rates of complete or partial remission ranging from 92% to 97%, compared to 81% in our study.11-14 Our results are consistent with other non-Mediterranean studies, with response rates ranging from 75 to 92%.15-17,19 Few studies reported the disease severity score, while in our study, most patients classified their disease as mild (42%); in other non-Mediterranean studies, most patients classified their disease as moderate (63% to 66%).16,17
Focusing on the genetic characteristics based on Mediterranean studies, five mutations (V726A, M694V, M694I, M680I, and E148Q) account for approximately 75% of FMF mutations. In our study, these mutations represent 86% of all known mutations. Seventeen patients (39%) were compound heterozygote. The most frequent gene mutation in our study was M694V, similar to both Mediterranean and non-Mediterranean studies. The most mutations we found in our population are classified as pathogenic/likely pathogenic according to the International Study Group for Systemic Autoinflammatory Diseases.4 The R408Q and P369S mutations are of uncertain significance and it is unclear if the E148Q mutation is a polymorphism or a disease-causing sequence alteration.
Even though MEFV mutations are more frequent in Mediterranean populations, the frequency of MEFV mutations is much higher in our cohort than in the Turkish general population, where M694V and P369S have frequencies of 2.6% (95% CI: 1.6-4.0) and 1.0% (95% CI: 0.5-2.0), respectively.27 The high yield of pathogenic mutations in suspected MEVF patients warrents genetic screening.
In conclusion, in a population of Dutch FMF patients, all originating from countries with a high FMF prevalence, the age of onset of symptoms and of diagnosis is similar to Mediterranean studies. Disease manifestations and genetic distribution of Dutch FMF patients is also comparable to those in Mediterranean regions. Our results suggest that environmental factors are of little influence on the clinical manifestations in FMF patients. Treating physicians in non-Mediterranean European countries should be aware of FMF in patients with a Mediterranean origin.
DISCLOSURES
All authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES