KEYWORDS
Hyperammonemia, OTC deficiency, urea cycle
BACKGROUND
Ammonia is continuously produced by the breakdown of protein. It easily crosses the blood-brain barrier and is toxic for the brain when plasma concentrations are increased (> 45 µmol/l), causing a wide range of symptoms (table 1).1,2 In the liver, ammonia is converted to water-soluble urea through the urea cycle after which it is excreted into the urine (figure 1). 
Increased plasma ammonia concentrations develop when the liver cannot fully convert ammonia. In approximately 90% of adult cases, this is caused by liver failure.3 However, if there are no signs of liver failure, non-hepatic causes of hyperammonemia should be investigated, including increased production or decreased metabolism of ammonia.4-8 When ammonia production exceeds the liver’s capability to convert this to urea, plasma ammonia levels rise. For example, gastrointestinal bleeding, urease-producing bacteria and urinary tract diversion can cause increased ammonia production.4,5
Another mechanism is portosystemic shunting, where ammonia bypasses the liver, preventing conversion to urea.6 Medication has also been associated with elevated ammonia levels; for example, valproic acid can lead to hyperammonemia.7,8 A rare cause of hyperammonemia in adults can be an inborn error of metabolism, e.g. hyperinsulinismhyperammonemia syndrome or urea cycle defects (UCD). Defects in the urea cycle may lead to high ammonia levels, especially in catabolic states. Although these defects are genetic primarily affecting children, the disorder does require medical attention in adulthood in most patients. Urea cycle defects present with typical changes in plasma amino acid levels, depending on the defective enzyme.9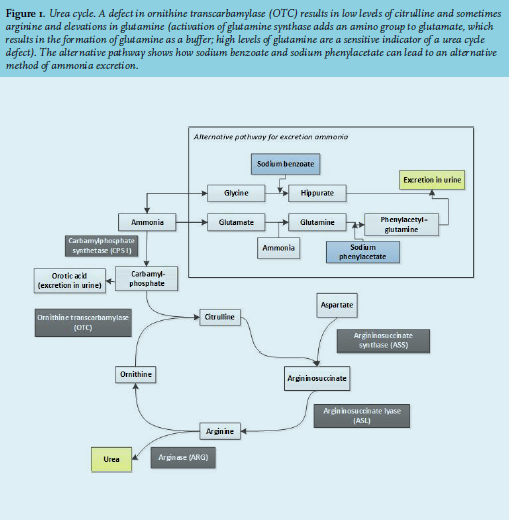
OBJECTIVE
The objective of this case report is to increase awareness for rare inborn errors of metabolism in adults and measure ammonia levels in patients with unexplained episodes of decreased consciousness. After excluding more frequent causes for hyperammonaemia, a urea cycle defect should be considered and appropriately diagnosed.
CASE REPORT
A 54-year-old female presented at the emergency department with a fever and a gradually decreasing level of consciousness over several days. Her medical history included borderline personality disorder, depression, alcohol and benzodiazepine addiction, a gastric bypass 20 years ago and a recent doxycycline-induced pancreatitis, which had led to severe weight loss. Upon physical examination, she had a pulse of 118 beats per minute, blood pressure of 100/60 mmHg, oxygen saturation of 90% and a temperature of 38.5 degrees Celsius. A chest X-ray showed an infiltration of the left lower lobe, which led to the diagnosis of pneumonia. She was treated with intravenous antibiotics and a silent delirium was hypothesized. However, despite adequate treatment of the infection, she did not regain full consciousness and exhibited manipulative and bizarre behavior. Consulting a neurologist and an additional neurological examination, including a computed tomography (CT)-cerebrum, did not reveal a cause. Flumazenil treatment, 0.25 mg intravenously, for suspected benzodiazepine intoxication led to temporary improvement, followed by discharge from the hospital. However, one month later, the patient was readmitted, again with a decreased level of consciousness. Her vital signs, physical and routine laboratory examination were normal this time. She had lost seven kilograms over several weeks and had become severely malnourished. Neurologic examination was normal aside from a decreased score of E3M5V5 on the Glasgow Coma Scale (GCS). Urinary drug screening was positive for benzodiazepines, but repeated treatment with flumazenil was unsuccessful. A CT scan and magnetic resonance imaging of the brain were normal. During admission, the patient exhibited the same bizarre behavior as during the previous admission. She became extremely agitated, spoke incoherently or tried to manipulate the staff. The psychiatrist was consulted but could not explain the symptoms by delirium, conversion disorder or psychosis and suggested a somatic cause for the symptoms. Extended chemical laboratory investigations showed an increased plasma ammonia level of 157 µmol/l (range 10-45 µmol/l). This hyperammonemia explained the symptoms including the bizarre behavior. Initial treatment with lactulose was started immediately, leading to insufficient improvement. Further examination was aimed at finding the underlying cause for the increased plasma ammonia. The differential diagnosis included an intrahepatic cause or portosystemic shunting, especially since the patient had a history of alcohol addiction. However, laboratory analysis, ultrasound and liver biopsy did not show any signs of liver cirrhosis or fibrosis. Portosystemic shunting was excluded with duplex ultrasound and CT scan. After excluding the above-mentioned most common causes for hyperammonemia, a potential defect in the urea cycle was suspected. A specialized center, Amsterdam University Medical Center (location AMC), was consulted and plasma amino acids and urinary orotic acid levels were measured. Glutamine levels were raised and citrulline and arginine levels were low (table 2). 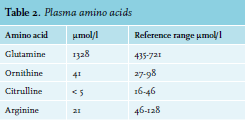
These levels could be caused by a defect in the urea cycle but also could have been caused by the severe malnutrition. In both cases, adequate amino acid supplementation is necessary, but high protein feeding can aggravate a urea cycle defect. Therefore, we decided to treat the patient as appropriate for a urea cycle defect with intravenous glucose, high carbohydrate feeding with adequate amino acid supplementation including arginine, but general protein restriction and sodium benzoate. Sodium benzoate works as a nitrogen scavenger by promoting an alternative pathway for the excretion of ammonia (see figure 1).10 After nourishment, laboratory tests showed persisting low citrulline levels and increased orotic acid in urine, pointing towards ornithine transcarbamylase (OTC) deficiency as the underlying cause of the hyperammonemia. Mutation analysis did not reveal a mutation in the OTC gene. The patient regained full consciousness, had a GCS score of E4M6V5 and could be discharged.
DISCUSSION
It is important to consider hyperammonemia when a patient has an unexplained altered conscious state. As discussed earlier, the most common cause is liver failure. If liver failure has been excluded, analysis for non-hepatic causes should be considered.11
Urea cycle defects are rare and usually present in childhood, with an estimated incidence of 2-3 in 100,000.12,13 However, 20% of patients present after 12 years of age.2 OTC is the most common urea cycle defect and is an X-linked disorder primarily affecting males. Presumably, as a result of random X-inactivation, clinical features in females may vary from severe episodes of hyperammonemia to asymptomatic disease.14 OTC deficiency may remain undiagnosed, especially in carrier females and can become apparent under extreme catabolic stress. In this particular case, the extreme weight loss of seven kilograms in several weeks after pancreatitis probably provoked the metabolic derangement. It is known that in up to 20% of all cases, no disease-causing mutation can be established.14 Therefore, plasma amino acids and urinary orotic acid analysis are crucial for confirmation of the diagnosis and to discriminate from other urea cycle defects.
In conclusion, hyperammonemia is strongly suspicious of a urea cycle defect in the absence of known hepatic disease. Since hyperammonemia and high glutamine levels may result in cerebral edema and even death, patients with high ammonia levels should be immediately treated as having a UCD, pending further diagnostic testing on samples that have been collected prior to treatment.
DISCLOSURES
All authors declare no conflicts of interest. C.E.M. Hollak is involved in pre-marketing research with Genzyme, Protalix and Idorsia.
REFERENCES