KEYWORDS
b thalassaemia, case report, congenital methaemoglobinaemia, CYB5R3
CASE PRESENTATION
A 61-year-old Ghanaian woman consulted her general practitioner (GP) because of dizziness, headaches, blurry vision and fatigue for three days. Low oxygen saturations of 78-80% upon physical examination made her GP refer her to the emergency department for further analysis.
The patient had a known medical history of well-controlled hypertension and non-insulin-dependent diabetes, for which she had used a thiazide diuretic, gliclazide and metformin for the past years, without recent changes. She did not smoke or use any intoxicating substances. Her symptoms occurred during work at a flower auction, where she had worked for decades. None of her colleagues experienced comparable symptoms. Her job did not involve direct contact with plants, flowers or soil.
The patient had returned from travelling to Ghana three weeks prior to presentation. While there she did not experience any symptoms. She did not use any malaria prophylaxis.
She was non-consanguineous, and had a sister who had died at the age of 30, but no cause was identified at autopsy. At the emergency department, she did not appear ill or dyspnoeic, with no signs of cyanosis. Measured vital signs are displayed in table 1. Further physical examination revealed no abnormalities. Arterial blood gas analysis was performed, the results are shown in table 1. Most notable is a significantly elevated methaemoglobin level at 24.9% (normal 1-2%).
The patient was admitted with a provisional diagnosis of acquired methaemoglobinaemia, thought to be caused by an unknown chemical compound possibly contracted during work. She was given 15 litres of oxygen and a single dose of 10 grams oral ascorbic acid (vitamin C). The Ministry of Social Affairs and Employment (Inspectorate SZW) was informed of the incident, as is required by Dutch law concerning (possible) severe workplace-related incidents.
During hospitalisation, the methaemoglobin levels declined in days to 13-14% but no further. Subsequently, methylene blue 1 mg/kg body weight was given intravenously, which resulted in a rapid and significant drop of the methaemoglobin level to 2.9%. The following day the methaemoglobin increased again to 4.8%, suggesting a congenital instead of an acquired cause of methaemoglobinaemia. As the patient was in a good clinical condition with normal oxygen saturation levels without supplemental oxygen, she was discharged.
At the outpatient clinic eight days later the patient reported no symptoms. Arterial blood gas analysis again showed high methaemoglobin levels (15.7%). Because of persistent methaemoglobinaemia in the absence of any apparent triggers, a congenital cause for the methaemoglobinaemia became more plausible. DNA sequence analysis was performed and revealed a novel homozygous variant in the CYB5R3 gene: NM 000398.6:c.181C>T, p.(Arg61Cys). This variant has not been described in the literature, and has only been found once in the 149,064 alleles of the Genome Aggregation Database. As other pathogenic variants1-3 are in close proximity to the variant found in our patient, we conclude that this variant is likely pathogenic and thus able to cause congenital methaemoglobinaemia.
With the patient’s natural methaemoglobin levels assumed to be around 15%, the sudden increase of methaemoglobin levels and accompanying symptoms as observed at presentation were concluded to be provoked by an unknown substance. Thorough evaluation of her medication, supplements and other products used did not result in a clear causative agent. The Inspectorate SZW could not identify any harmful chemical compounds at her workplace. Reviewing the literature on CYB5R3 deficiency it struck us that it is usually accompanied by a reactive polycythaemia as compensatory mechanism.4-6 As our patient had normal haemoglobin levels and a low mean corpuscular volume, we performed additional tests demonstrating increased HbF and lowered HbA levels (table 1). Genetic analysis revealed a delta-beta thalassaemia (deletion exon 3-9 HBBP1 gene). Thus, our patient was found to have congenital methaemoglobinaemia caused by a novel homozygous variant, in conjunction with a heterozygous delta-beta-thalassaemia, resulting in normal non-physiological haemoglobin levels.
At follow-up, the patient was instructed to abstain from drugs, as some may provoke symptomatic methaemoglobinaemia. She was advised to consult her GP or the emergency department immediately if her symptoms relapsed. Her GP was informed of potential harmful medications and supplements (including sulphonamides, metoclopramide and nitrates) for future reference. An overview of all substances capable of inducing methaemoglobinaemia can be found at www.uptodate. com/contents/clinical-features-diagnosis-and-treatment-ofmethemoglobinemia.7 The combination of these diagnoses has no clinical consequences for the patient. Genetic counselling and testing was offered to her direct family members, including pre-conception counselling for her offspring.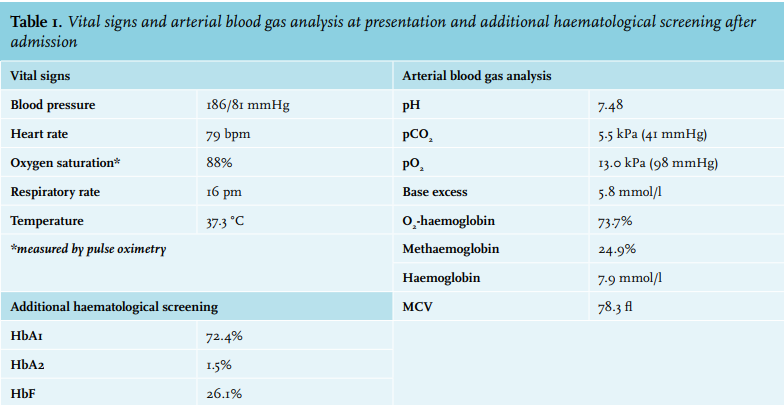
DISCUSSION
Methaemoglobinaemia is a rare condition where blood levels of methaemoglobin are elevated. Methaemoglobin carries ferric (Fe3+) instead of ferrous (Fe2+) iron on haemoglobin molecules, hampering binding of oxygen, causing a cyanotic state.4,8 Causes of methaemoglobinaemia can be acquired or congenital. Acquired causes are the most prevalent, as various oxidising agents (including certain medications and drugs) are able to increase oxidation activity of ferrous to ferric iron,8 resulting in an acute methaemoglobinaemia.
Treatment is based on severity of symptoms and methaemoglobin levels. The administration of 1-2 mg/ kg body weight methylene blue can adequately reduce methaemoglobin to normal levels.9 In addition, supplemental high-flow oxygen is effective in elevating plasma oxygen levels, subsequently stimulating conversion of methaemoglobin to haemoglobin.4 Previously, ascorbic acid (vitamin C) was considered an effective treatment,10 but oral administration does not appear to result in therapeutic plasma concentrations.11,12
Most congenital causes of methaemoglobinaemia are based on genetic defects in cytochrome b5 reductase (CYB5R), occurring in almost all ethnic groups. CYB5R deficiency can be expressed in erythrocytes only (type 1), or in all tissues (type 2).8 Patients with type 1 CYB5R deficiency have persistent pallor or cyanosis, or may remain asymptomatic.13 In contrast, type 2 CYB5R deficiency results in failure to thrive, severe neurological symptoms and mental retardation.14,15 Patients with chronically elevated levels of methaemoglobin often have a compensatory polycythaemia.4-6
Our patient’s first presentation at age 61, together with the apparent acute onset and lack of family history, suggested an acquired cause of her methaemoglobinaemia. While admitted, stabilisation of the methaemoglobin levels to around 14% and absence of possible toxins implied a congenital cause. The observed variant and the absence of symptoms earlier in life, suggest a type 1 CYB5R deficiency in our patient. Heterozygous delta-beta thalassaemia normally leads to low-normal haemoglobin levels or mild anaemia. The combination of type 1 congenital methaemoglobinaemia and carriership of delta-beta thalassaemia resulted in normal haemoglobin levels in our patient. 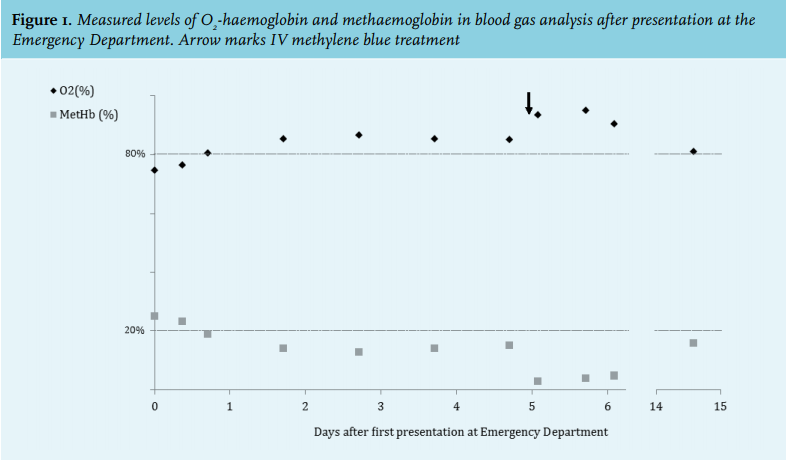
CONCLUSION
The described combination of two congenital erythrocyte abnormalities has to our knowledge only been reported once, in newborn twins.16 Our patient did not experience any symptoms related to one or both of the congenital defects until older age. We hypothesise that the combination of both defects with the subsequent adjusted ‘normal’ haemoglobin levels resulted in these abnormalities going undetected.
DISCLOSURES
This case was presented at the Dutch Acute Medicine Congress in Enschede, the Netherlands, on 10 June 2016. All authors report no potential conflicts of interest.
REFERENCES