
INTRODUCTION
Hyponatraemia, defined as a serum sodium concentration of < 135 mmol/l, is the most frequently encountered electrolyte disorder in clinical practice.1,2 The syndrome of inappropriate antidiuretic hormone secretion (SIADH) is the most common underlying cause and can be suspected in euvolaemic patients with hyponatraemia, decreased effective osmolality (< 275 mOsm/kg), high urine osmolality (> 100 mOsm/kg of water) and high urine sodium levels (> 40 mmol/l).2-4 SIADH should only be diagnosed after exclusion of adrenal, thyroid and pituitary insufficiency.2,4 However, the evaluation of these contains certain pitfalls. To emphasise the importance of correctly excluding adrenal and pituitary insufficiency, we present two cases in which a suspected SIADH was actually caused by hypopituitarism.
CASE REPORT 1
A 74-year-old male patient with no medical history was referred because of hyponatraemia. In the last three months the patient experienced progressive fatigue, weakness, weight loss and recurrent collapses. At the emergency department, a hypotonic hyponatraemia of 128 mmol/l with a urine sodium level of 63 mmol/l and urine osmolality of 671 mOsm/kg were measured. Thyroid-stimulating hormone levels were within the normal range (1.9 mIU/l) after which SIADH was considered the most likely diagnosis. Further evaluation was scheduled at the outpatient clinic.
At presentation, the patient was euvolaemic with a blood pressure of 111/75 mmHg and a pulse rate of 77 bpm. Explicit paleness was present and additional testicular examination revealed an estimated reduced volume of 10 ml. Based on these symptoms and the unexplained hyponatraemia, we suspected adrenal insufficiency. Additional testing revealed hypocortisolism with low-normal ACTH levels, hypogonadotropic hypogonadism, central hypothyroidism and mild hyperprolactinaemia (table 1). Cranial MRI showed a 15 mm lesion in the sella turcica suggestive of a Rathke’s cleft cyst. (figure 1). Based on these findings we established the diagnosis of panhypopituitarism due to a Rathke’s cleft cyst. After starting replacement therapy with hydrocortisone, levothyroxine and testosterone this patient quickly made a full recovery. Ultimately, the sodium levels normalised to 140 mmol/l. A follow-up MRI scan six months later showed no growth of the Rathke’s cleft cyst. 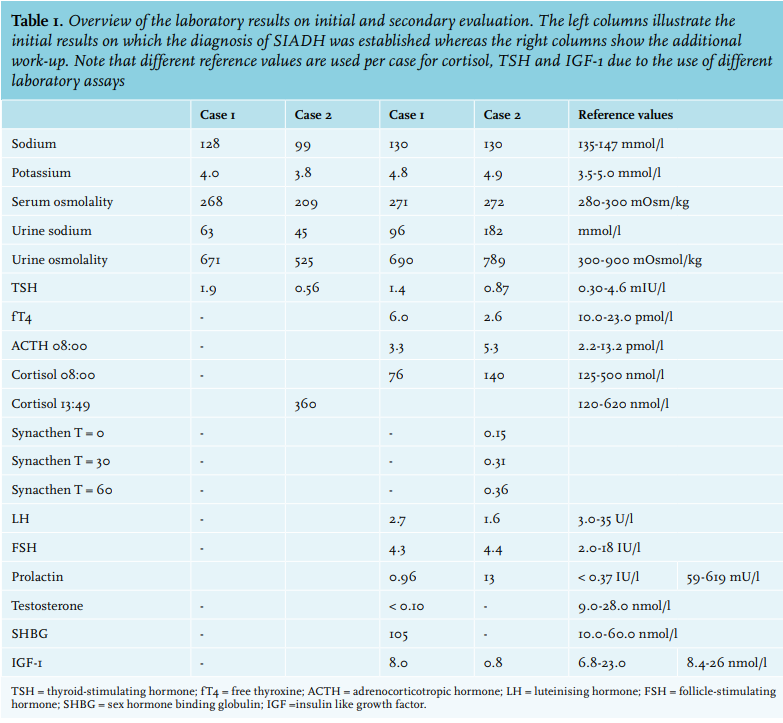
CASE REPORT 2
A 53-year-old Bolivian female patient was admitted to the intensive care unit with severe pneumonia and hypotonic hyponatraemia of 99 mmol/l. She had experienced general weakness for two weeks. Urine sodium was 45 mmol/l and urine osmolality was 525 mOsm/kg. Random cortisol was 360 nmol/l and thyroid-stimulating hormone was 0.56 mIU/l. Therefore, SIADH secondary to pneumonia was diagnosed and the patient was treated with antibiotics, hypertonic saline and water restriction. At discharge, she had fully recovered and her sodium level was 134 mmol/l. One week later she was readmitted with progressive abdominal pain, nausea, diarrhoea and prolonged weakness. On examination she was pale and hypotensive (BP 97/53 mmHg, pulse 72 bpm). Blood tests revealed recurrent hypotonic hyponatraemia of 130 mmol/l with a urine osmolality of 789 mOsm/kg and urine sodium of 182 mmol/l (table 1).
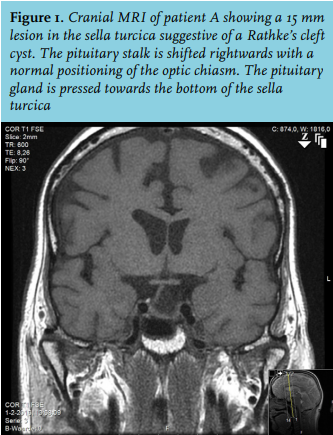
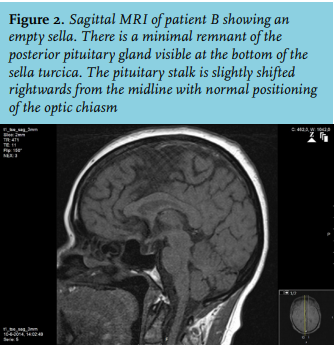
Based on the history, hypotension and recurrent hyponatraemia, further testing was performed. A morning cortisol level was 140 nmol/l and the ACTH stimulation test showed an insufficient response. Furthermore, central hypothyroidism and growth hormone deficiency were present (table 1). Cranial MRI showed an empty sella (figure 2). Additional history taking revealed that her last pregnancy in Bolivia had been complicated by heavy blood loss at delivery for which she received multiple blood transfusions. After this delivery she was unable to provide breastfeeding and she no longer had menstruation. Based on these findings we concluded a panhypopituitarism due to Sheehan’s syndrome. The patient quickly recovered after starting replacement therapy with hydrocortisone and levothyroxine.
DISCUSSION
In the majority of hospitalised patients with hyponatraemia, SIADH is the primary cause.1-3 Underlying reasons for SIADH are diverse but can be subdivided in malignant diseases, pulmonary disorders, central nervous system disorders and the use of certain types of drugs.2,3 In SIADH, a physiological base (e.g. volume depletion of the extracellular fluid or hypertonicity) for the secretion of ADH is lacking, subsequently leading to inappropriately elevated ADH levels.3,4
In contrast, hypopituitarism with secondary adrenal insufficiency is a much rarer cause of hyponatraemia. Space-occupying lesions, trauma or infections in the pituitary gland as well as genetic disorders involved in pituitary development can lead to corticotrophin deficiency.5,6 As endogenous glucocorticoids suppress both ADH and corticotrophin releasing hormone release in the hypothalamus, glucocorticoid deficiency leads to elevated ADH levels.7,8 These elevated ADH levels actually mimic SIADH. Hence, distinguishing these two clinical conditions might be difficult, especially in the absence of hypovolaemia or dehydration.8
Diederich reviewed 28 cases of severe hyponatraemia due to hypopituitarism with secondary adrenal insufficiency. Scanty/missing axillary or pubic hair (85.7%), nausea or vomiting (75%), a pale, doughy skin (71.4%) and in males a testicular volume of less than 12 ml (71.4%) were the most common clinical signs.9 Furthermore, anorexia, weight loss, fatigue and lack of energy or stamina are clinical signs of glucocorticoid deficiency often seen in adrenal insufficiency.5
The first pitfall described in our cases is the evaluation of thyroid function by measurement of solely TSH. Central hypothyroidism was present in both cases as free thyroxine levels were decreased and TSH was inappropriately normal. Even though hypothyroidism-induced hyponatraemia is rare and might only occur in severe hypothyroidism, the presence of central hypothyroidism could have pointed towards a central endocrinological problem at an earlier stage.10 Therefore, measuring solely TSH is not sufficient for excluding central endocrinological disorders in suspected SIADH.
Another pitfall is the exclusion of hypocortisolism by randomly measured cortisol levels. As serum cortisol levels are highest in the early morning, a cortisol level of < 100 nmol/l is strongly suggestive of adrenal insufficiency whereas a level of > 500 nmol/l excludes adrenal insufficiency.5,11 These cut-off values should be interpreted in the clinical context and are variable due to the use of different cortisol assays.12 Case 2 illustrates that randomly measured cortisol levels are of limited use. As clinical suspicion is high and a basal cortisol level is inconclusive, dynamic testing should be performed. Both the insulin tolerance test and the short synacthen test have been validated for diagnosing adrenal insufficiency.5,11,12 However, since the adrenal cortex might still be responsive in the first four weeks after onset of ACTH deficiency, the short synacthen test could be falsely normal.5,11,12
CONCLUSION
Hypopituitarism with secondary adrenal insufficiency is a rare cause of hyponatraemia. It can be difficult to distinguish from SIADH, especially in the absence of hypovolaemia or dehydration. In any suspected SIADH, a morning cortisol, TSH and free thyroxine should be included in the initial work-up. If basal morning cortisol levels are inconclusive, dynamic testing of adrenal function should be performed before establishing the diagnosis SIADH.
DISCLOSURES
All authors declare no conflict of interest. No funding or financial support was received.
REFERENCES