DIAGNOSIS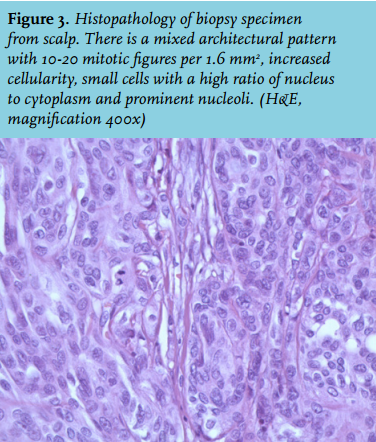
A soft-tissue sarcoma, bone metastasis of an unknown distant primary carcinoma or lymphoma was initially considered. Magnetic resonance imaging revealed a large heterogeneous tumour located both intracranially and extracranially with mass effect. The skull was apparently intact. The differential diagnosis was not revised. A histological biopsy (figure 3) showed an atypical meningioma (WHO grade II). Palliative treatment consisted of resection of the extracranial part of the tumour followed by 60 Gy radiotherapy in 30 fractions on the intracranial part. During surgery direct invasion of the skull was noted by the neurosurgeon. Figure 4 shows the situation shortly after treatment. Considering the palliative intentions of the treatment, the preference and the age of the patient, no regular follow-up imaging or ambulatory visits were planned. Twelve months later the patient developed a left-sided hemiparesis and dysarthria. A CT scan showed a mass suggestive of residual/relapse meningioma. No further extracranial growth was observed. After extensive consideration of diagnostic and/or therapeutic options with the patient and his family, no further procedures were performed besides starting dexamethasone. He was entrusted into care of his general practitioner. Six months later we were informed the patient had died at home.
Meningiomas are tumours arising from the dural coverings of the brain and they are the most common primary intracranial tumour. Most meningiomas remain asymptomatic throughout life and are only found incidentally with imaging or at autopsy. Their rate of growth is typically slow. The prevalence of meningiomas found at autopsy in persons over 60 years of age is 3%, and the majority of the lesions are less than 1 cm in diameter.1 However, progressive enlargement of the tumour can lead to focal or generalised seizure disorders or neurological deficits caused by compression of adjacent neural tissue. The aetiology of meningioma is not well understood, although there are several recognised risk factors. The incidence is highest after the fifth decade of life. Meningiomas are twice as common in the female as in the male population. Progesterone receptors are expressed in many meningiomas and some also express oestrogen receptors. Furthermore pregnancy and menopause have been associated with an increased incidence of meningiomas suggesting an aetiological role for hormones. Cranial irradiation for intracranial tumours or disease prophylaxis has also been related to later-onset meningioma. Neurofibromatosis type 2 (NF2) is an autosomal dominant disorder due to a mutation in a tumour suppressor gene on chromosome 22, predisposing to multiple neoplastic lesions. Approximately half of the individuals with NF2 have meningiomas, often at a young age and multiple meningiomas are often present. The meningiomas seen in patients with NF2 are more frequently atypical or anaplastic compared with sporadic tumours. In addition, up to 60% of sporadic meningiomas show a somatic mutation of NF2 gene. Meningiomas can arise anywhere from the dura, most commonly within the skull and at sites of dural reflection (falx cerebri, tentorium cerebelli, venous sinuses). Other less common sites include the optic nerve sheath and choroid plexus; approximately 10% arise in the spine. Very rarely, meningiomas can arise at extradural sites.
Histological grading of meningiomas is based on the WHO classification (table 1). Most (about 90%) are WHO grade I, reflecting their benign nature. Atypical meningiomas (WHO grade II) make up 5-7% and anaplastic variants (WHO grade III) 1-3%. Patients with WHO grade II or grade III meningiomas are more likely to have invasive disease, a local recurrence following the initial treatment, and ultimately to have a shorter overall survival compared with patients with a WHO grade I meningioma. Individual management of patients with a meningioma depends on the signs and symptoms, age, WHO grade, site and size of the tumour. Most patients with small asymptomatic meningiomas can be safely observed. Symptomatic meningiomas and asymptomatic tumours that are expanding, infiltrating, or associated with surrounding oedema should be surgically resected if feasible. Radiation therapy can be used after incomplete resection, after recurrence and when tumour histology reveals atypia or anaplasia.2 Extracranial meningiomas are rare and the majority have a secondary extension of the primary intracranial tumour through the skull, without radiological evidence of osteolysis.3
In conclusion, meningiomas are mostly benign intracranial lesions and extracranial extension of atypical meningioma is rare. Management should be tailored depending on the patients characteristics and wishes and difficulties in decision-making may arise.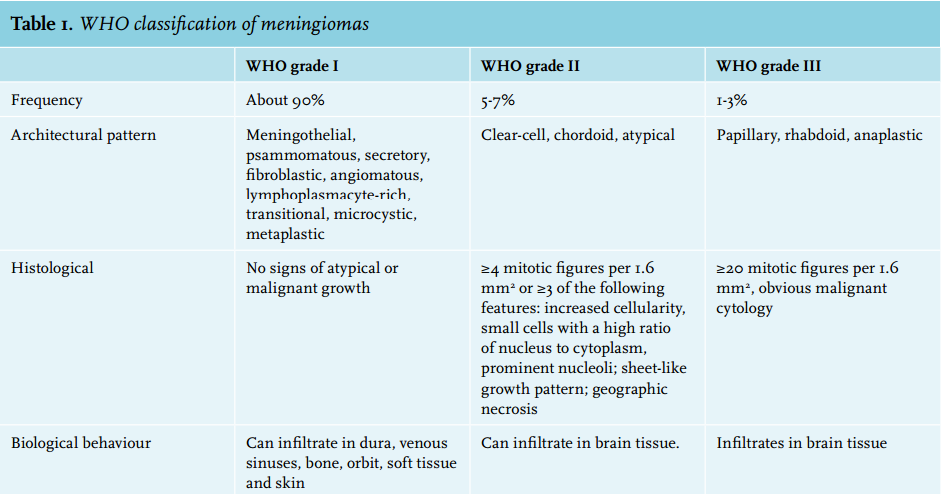
REFERENCES